
Abstract
SUMMARY: Morning glory syndrome is characterized by a congenital optic disc defect that resembles the eponymous flower. We present the MR imaging findings of 2 pediatric patients with morning glory disc anomaly and persisting embryonal infundibular recess, another rare malformative finding, a previously unreported association. Neuroradiologists should be aware of the possible presence of a persisting embryonal infundibular recess in patients with morning glory syndrome, to aid in the differential diagnosis including other pituitary malformations such as pituitary stalk duplication.
ABBREVIATIONS:
- MGDA
- morning glory disc anomaly
- MGS
- morning glory syndrome
- PEIR
- persisting embryonal infundibular recess
The morning glory disc anomaly (MGDA) is a rare, congenital ocular defect diagnosed by fundoscopy. It is characterized by a funnel-shaped macropapilla with central neuroglial remnants, surrounded by an elevated, pigmented chorioretinal ring, named after its resemblance to the morning glory flower.1,2 While it is usually unilateral, bilateral MGDA may occur.3 Even though it may be an isolated finding, it can present in the context of a morning glory syndrome (MGS) when associated with other congenital abnormalities, including craniofacial, skull base, optic pathway, corpus callosum, vascular, and pituitary ones.2,4
The embryonal infundibular recess is a funnel-shaped liquoral space of the third ventricle floor extending into the pituitary stalk, normally obliterated during development, whose persistence is defined as persisting embryonal infundibular recess (PEIR). It has been rarely reported5,6 and appears as an unusual expansion of the third ventricle floor into the sella turcica with loss of the normal recess contours. At MR imaging, it appears as an intrasellar cyst, communicating with the third ventricle. The sella turcica may be expanded, and the pituitary gland is typically thinned (empty sella).
We present 2 patients with fundoscopically confirmed MGDA whose brain MR imaging showed several of the above-mentioned anomalies as well as a PEIR. The association between MGDA and PEIR has not been reported currently, to the best of our knowledge.
Case Series
Patient 1.
This 3-year-old boy presented with short stature and migraine. Fundoscopy revealed a MGDA of the right eye. Several stimulus tests confirmed a complete growth hormone isolated deficiency.
MR imaging showed hypophyseal hypoplasia (Fig 1) and a stubby, thickened, and inferiorly dropped optic chiasm with normal signal intensity (Fig 2). Sagittal images also showed a dysmorphic hypothalamic infundibulum and pituitary stalk. Indeed, there was a direct communication between the third ventricle and the sellar cavity, suggesting a PEIR (Fig 1). The acquisition of a constructive interference in steady state sequence confirmed this finding, better depicting a tubular morphology of the pituitary stalk in the absence of sphenoidal meningocele (Fig 1). The sella was mildly enlarged, and there was clival hypoplasia. Additional findings included a corpus callosum body and splenium partial agenesis and a small interhemispheric arachnoid cyst (Fig 1).

Sagittal contrast-enhanced T1-weighted (B) and high-resolution T2-weighted (D) images with corresponding stylized anatomic drawings (A and C) of patient 1 (upper row) and patient 2 (lower row) depicting the main findings reported in the text, including PEIR.

Coronal T2-weighted images of patient 1 (A) and patient 2 (B) showing thickening of the optic chiasm (arrows).
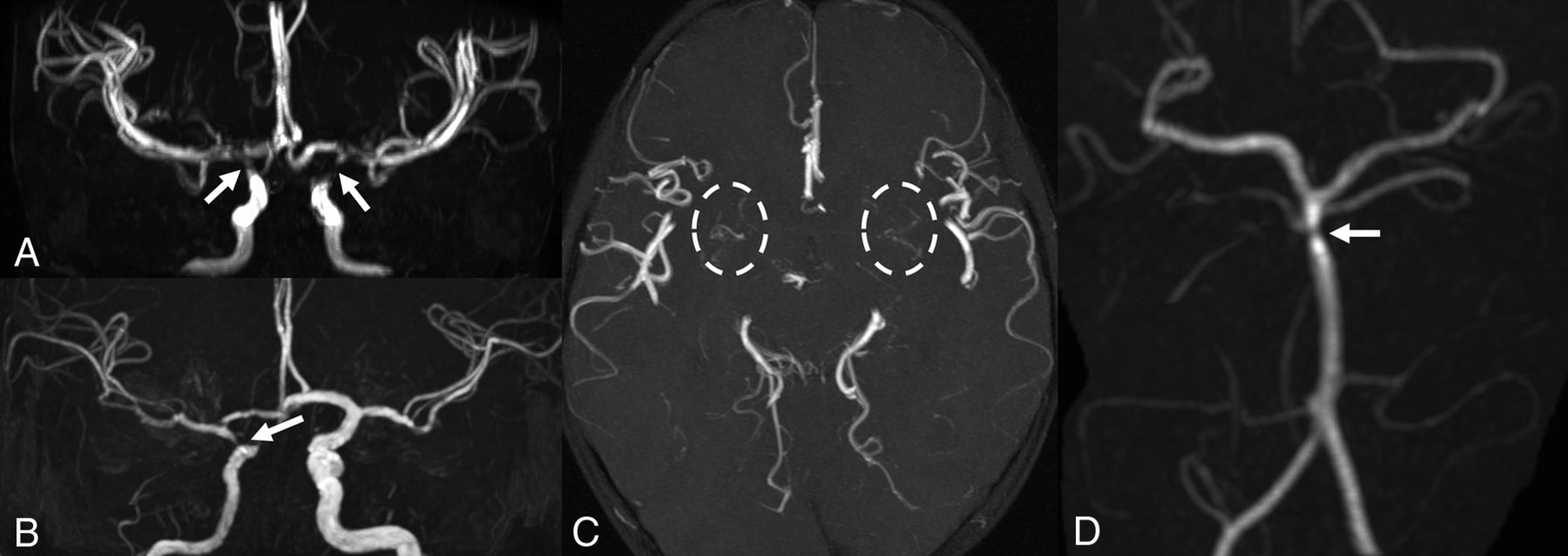
Finally, bilateral supraclinoid ICA and M1 MCA segment narrowing was detected on MRA, with thin collateral lenticulostriate vessels, suggestive of Moyamoya syndrome (Fig 3). Due to these findings and the history of headache, a DSC PWI study was performed, indicating a preserved cerebrovascular reserve capacity.
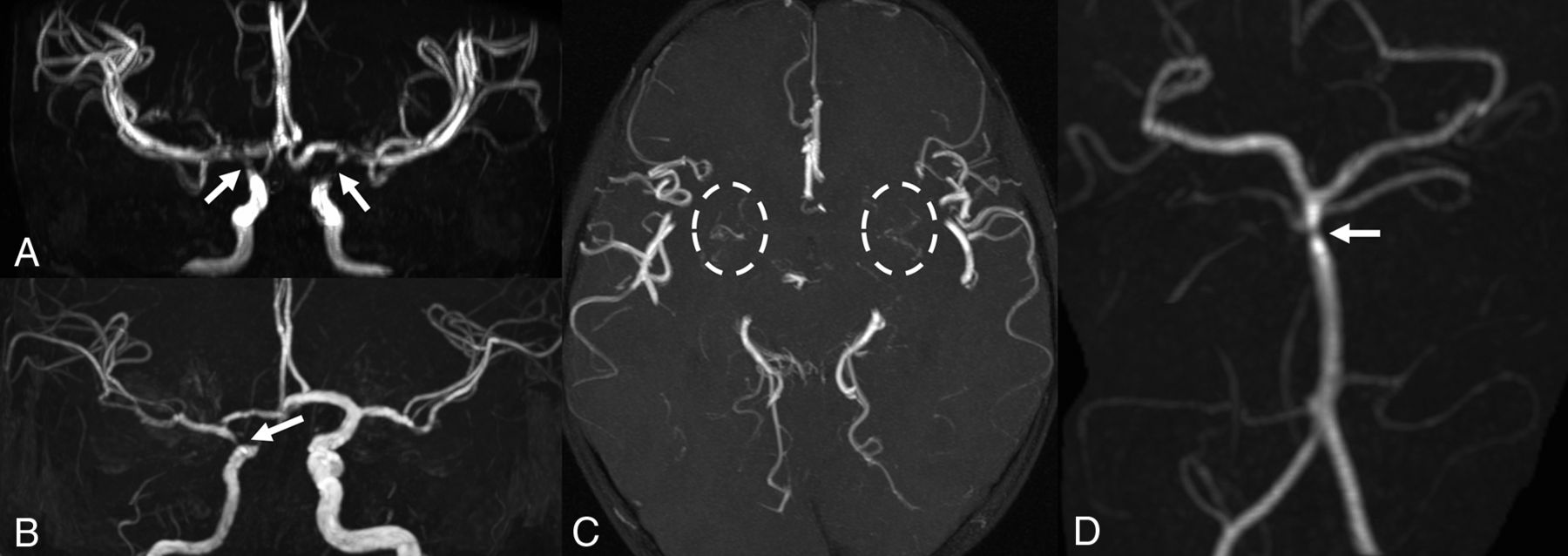
MR angiography. Patient 1 (A and C) has bilateral focal stenoses of the internal carotid arteries (arrows) with thin collateral vessels in the lenticulostriate regions (dashed circles). Patient 2 shows unilateral stenosis of the right internal carotid (B) and basilar (D) arteries (arrows).
Genetic testing did not identify any PAX6 gene mutations, encoding a transcriptional regulator involved in lens placode formation, previously reported in several ocular anomalies, including MGDA.7
Patient 2.
This 13-year-old boy came to our attention for short stature and chronic headache. Laboratory tests confirmed growth hormone deficiency.
Brain MR imaging showed sellar enlargement, adenohypophysis hypoplasia, and PEIR (Fig 1). The last finding was better defined on a sagittal driven equilibrium sequence, which demonstrated an evident communication between the third ventricle and sella turcica. The infundibular recess was, in fact, absent, and consequently, the pituitary stalk had a tubular appearance with thin walls.
The right part of the optic chiasm and the ipsilateral prechiasmatic optic nerve were displaced slightly downward. On the same side, partial hypoplasia of the sphenoid body with a small meningoencephalocele was found (Fig 2). A thin posterior defect of the right ocular globe was also noted. The corpus callosum was uniformly slightly thickened (according to Garel et al8), but not clearly dysplastic.
MRA detected right ICA hypoplasia with severe supraclinoid narrowing and distal basilar artery stenosis (Fig 3). DSC-PWI showed only mild elongation of right frontal lobe MTT. This, associated with MRA findings, prompted yearly follow-up MR imaging which, during a 3-year period, did not show progression.
On the basis of these findings, MGS was suspected, and the patient underwent an ophthalmologic evaluation, confirming an MGDA.
Discussion
MGDA is a sporadic condition, but it has been previously described as an isolated finding in a mother and child.9 Our patients were initially referred for pituitary MR imaging due to growth hormone deficiency, revealing a PEIR. The pituitary stalk was apparently duplicated on coronal images because of its tubular morphology and direct communication with the third ventricle. This finding should be carefully evaluated to avoid a potential misdiagnosis of other pituitary stalk malformative conditions, such as duplication.2 The anomalous infundibulum perforation, in the absence of meningoencephaloceles, was clearly defined by high-resolution volumetric T2-weighted sequences (CISS and driven equilibrium in our cases). To the best of our knowledge, among reported MGDA cases in the literature, none had associated PEIR.
Among the possible midline abnormalities reported in MGS, there is the craniopharyngeal canal, not present in our patients.10 It represents a rare, well-corticated midline channel of the sphenoid body that develops from the sellar floor to the nasopharynx. Its origin has been hypothesized from incomplete closure of the Rathke pouch, the precursor of the adenohypophysis. This entity can be variably associated with encephalocele and hypopituitarism, most likely due to ectopic adenohypophysis and even tumors (gliomas, dermoids, teratomas, craniopharyngiomas, or adenomas).11
Both of our patients had chiasmatic abnormalities with thickening and inferior drooping. On the basis of our experience and the lack of progression on follow-up imaging, we agree with Doneda et al12 on the dysplastic and not neoplastic nature of the chiasmal thickening. In our second patient, in the absence of sellar floor defects, chiasmatic prolapse was related to ipsilateral sphenoidal hypoplasia, which could be considered the less severe form of a sphenoid malformative spectrum ending with a meningoencephalocele.
While corpus callosum partial agenesis has already been described in MGS,2 global thickening, as observed in the second case, though in the absence of dysmorphisms or psychomotor delay, has never been reported.
Intracranial vascular abnormalities in MGS, not necessarily ipsilateral to the optic disc anomaly, could be due to the close relationship of the ICA and optic vesicle development during the fourth week of gestation.1,4,12 Arterial involvement, most commonly affecting the anterior circulation, ranges from mild hypoplasia to focal stenosis or complete agenesis and progressive Moyamoya syndrome.3,13 Arterial alterations in our patients were not associated with cerebrovascular reserve impairment. While it is not possible to completely exclude Moyamoya syndrome without more prolonged monitoring, we believe a nonprogressive arterial dysplasia is a more probable diagnosis. We recommend PWI in addition to MRA and follow-up for MGS-associated vasculopathy evaluation.
Conclusions
While different craniofacial, pituitary, optic, vascular, and cerebral midline anomalies have been previously reported in association with MGDA, our patients are the first in whom PEIR has also been found. Neuroradiologists should be aware of the possible presence of PEIR in patients with MGS, to aid in the differential diagnosis including other pituitary malformations such as pituitary stalk duplication. We also suggest the use of high-resolution T2-weighted sequences on the sagittal plane to better define the infundibulum in case of abnormalities.
Acknowledgments
The authors thank Rosanna Cianniello for the anatomic drawings.
REFERENCES
- Received October 22, 2018.
- Accepted after revision January 14, 2019.
- © 2019 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}