
Abstract
SUMMARY: Oculodentodigital dysplasia, a rare genetic disorder caused by mutations in the gene encoding gap junction protein 1, classically presents with typical facial features, dental and ocular anomalies, and syndactyly. Oligosymptomatic patients are common and difficult to recognize, in particular if syndactyly is absent. Neurologic manifestation occurs in approximately 30% of patients, and leukodystrophy or T2 hypointensity of gray matter structures or both have been noted in individual patients. To investigate MR imaging changes in oculodentodigital dysplasia, we retrospectively and systematically reviewed 12 MRIs from 6 genetically confirmed patients. Diffuse supratentorial hypomyelination, T2-hypointense Rolandic and primary visual cortex, and symmetric involvement of middle cerebellar peduncle, pyramidal tract, and medial lemniscus was present in all, T2-hypointense pallidum and dentate nucleus in 2 patients each. This consistent, characteristic pattern of diffuse supratentorial hypomyelination and brain stem involvement differs from other hypomyelinating and nonhypomyelinating leukodystrophies with brain stem involvement, and its recognition should trigger genetic testing for oculodentodigital dysplasia.
ABBREVIATIONS:
- NAWM
- normal appearing white matter
- ODDD
- oculodentodigital dysplasia
Oculodentodigital dysplasia (ODDD) is a rare genetic disorder caused by mutations in the gene encoding gap junction protein 1, also called Connexin 43 (GJA1; Online Mendelian Inheritance in Man Nos. 164200 and 257850). Patients show typical facial features including a narrow nose, hypoplastic alae nasi, short palpebral fissures, and epicanthal folds (92% of patients). Ocular, in particular anterior chamber, anomalies, and dental involvement, namely microdontia and enamel hypoplasia (approximately 70%), as well as syndactyly of fourth and fifth finger (in 80%) are common.1 Neurologic manifestations occur in approximately 30% of patients, including spastic paraparesis, prominent urinary incontinence, ataxia, dysarthria, seizures, and, infrequently, developmental delay or mild intellectual disability.1,2
Beighton et al3 first reported CT with calcification of the basal ganglia in patients with ODDD in 1979. T2 hypointensity of deep gray matter structures and supratentorial cortical regions (eg, the central region) was subsequently reported for individual patients examined with MR imaging. Diffuse T2 hyperintensity of supratentorial white matter has been observed in individual cases, brainstem involvement in three.4⇓–6 Having noted brain stem involvement in our patients, we retrospectively reviewed cranial MR imaging, including 1H-MR spectroscopy and DWI, of 6 genetically confirmed patients with MR imaging seen at our institutions to investigate the pattern of MR imaging changes in ODDD.
Materials and Methods
We retrospectively identified 6 patients with ODDD from 4 families with previous MR imaging who were diagnosed and/or followed at University Hospital Heidelberg and VU University Medical Center and Amsterdam Neuroscience. Their 12 cranial MRI scans (age at examination, 5–52 years; mean/median, 25.9/18.7 years) were systematically reviewed in consensus by a neuroradiologist (I.H.) and pediatric neurologist (N.I.W.), each with >15 years of experience in assessing white matter diseases. The study was approved by Heidelberg University (S-760/2018). Axial T2WI and T1WI were available for all 12 MRI scans, and sagittal T1WI/T2WI and DWI with ADC, for at least 1 MRI per patient (11/12 and 9/12 MRIs, respectively). MR imaging was assessed for the extent of T2WI white matter changes (focal/diffuse), corresponding T1WI signal changes, and involvement of specific white matter structures, namely the corpus callosum, anterior and posterior limbs of the internal capsule, pyramidal tract, brain stem tracts, and cerebellar peduncles. The central region and primary visual cortex, pallidum, thalamus, substantia nigra, red nucleus, and dentate nucleus were visually compared with normal-appearing white matter (NAWM) and with contrast in age-matched controls.
Gradient-echo T2* and SWI, available for 4 patients (6 MRIs: patient 1 [last MRI], patient 2, patient 5, patient 6), were checked for T2* hypointensities due to calcifications and were compared with contrast in age-matched controls. DWI and ADC maps were inspected for areas of altered diffusion compared with surrounding NAWM or gray matter. Quantitative 1H-MR spectroscopy of the supratentorial white matter was acquired in all patients (point-resolved spectroscopy sequence or STEAM localization, TE = 20–35 ms). Metabolite values were quantified using LCModel (http://www.lcmodel.com/),7 compared with control values for each scanner,8⇓–10 and graded as normal (within the mean ± 1 SD of controls), high/low-to-normal (differing by 1–2 SDs from the mean of controls), and high/low (above/below the mean ± 2 SDs).
Patients
Diagnosis was confirmed by mutation analysis of GJA1 in all patients; genetic and MR imaging findings are summarized in On-line Table 1.
Patient 1 had had correction of bilateral syndactyly of D IV and V at 9 months and was evaluated for action and intention tremor at 4 years of age. He also showed myopia, astigmatism, and hypodontia. His father (patient 5), older sister, paternal uncle, and grandmother (no MRIs) also had syndactyly of D IV and V partly in association with ocular and dental findings. Patients 2 and 6, adolescent daughter and mother, had bilateral syndactyly corrected in childhood; spastic paraparesis and urinary incontinence had developed in the mother in her mid-30s.11,12 Patient 3 had mild motor delay. At 6 years of age, neurogenic bladder was diagnosed with secondary incontinence and frequent bouts of cystitis. Neurologic examination revealed mild spasticity and posterior column signs. Patient 4 had syndactyly of D IV and V of her right hand. She had normal motor milestones but was always clumsy. Motor symptoms became more prominent around 40 years of age with mild spastic paraparesis, ataxia, and posterior column signs on examination. She also developed neurogenic bladder. Optic discs were pale, pursuit movements saccadic.
MR Imaging
All patients had diffuse, mild T2 hyperintensity of the supratentorial white matter with normal T1 signal, characteristic of hypomyelination (Fig 1 and On-line Fig 1). Three patients had additional, focally more pronounced T2 hyperintensity with corresponding T1 hypointensity. The corpus callosum was involved in all patients, varying from T2 hyperintensity of the lateral splenium in the last MR imaging of patient 1 at 8.9 years of age to an entirely T2 hyperintense corpus callosum (patient 4). On sagittal images, the corpus callosum was thin in patients 2–6. The posterior limb of the internal capsule was T2 hyperintense in all patients and MRIs, while the anterior limb was normal in the youngest 2 and was involved to a varying extent in the older 4 patients. The younger 3 patients imaged within the first 2 decades had some subcortical NAWM, while this was scarce (patient 5) or absent (patients 4 and 6) in the older patients imaged at 41–52 years of age (On-line Fig 1).
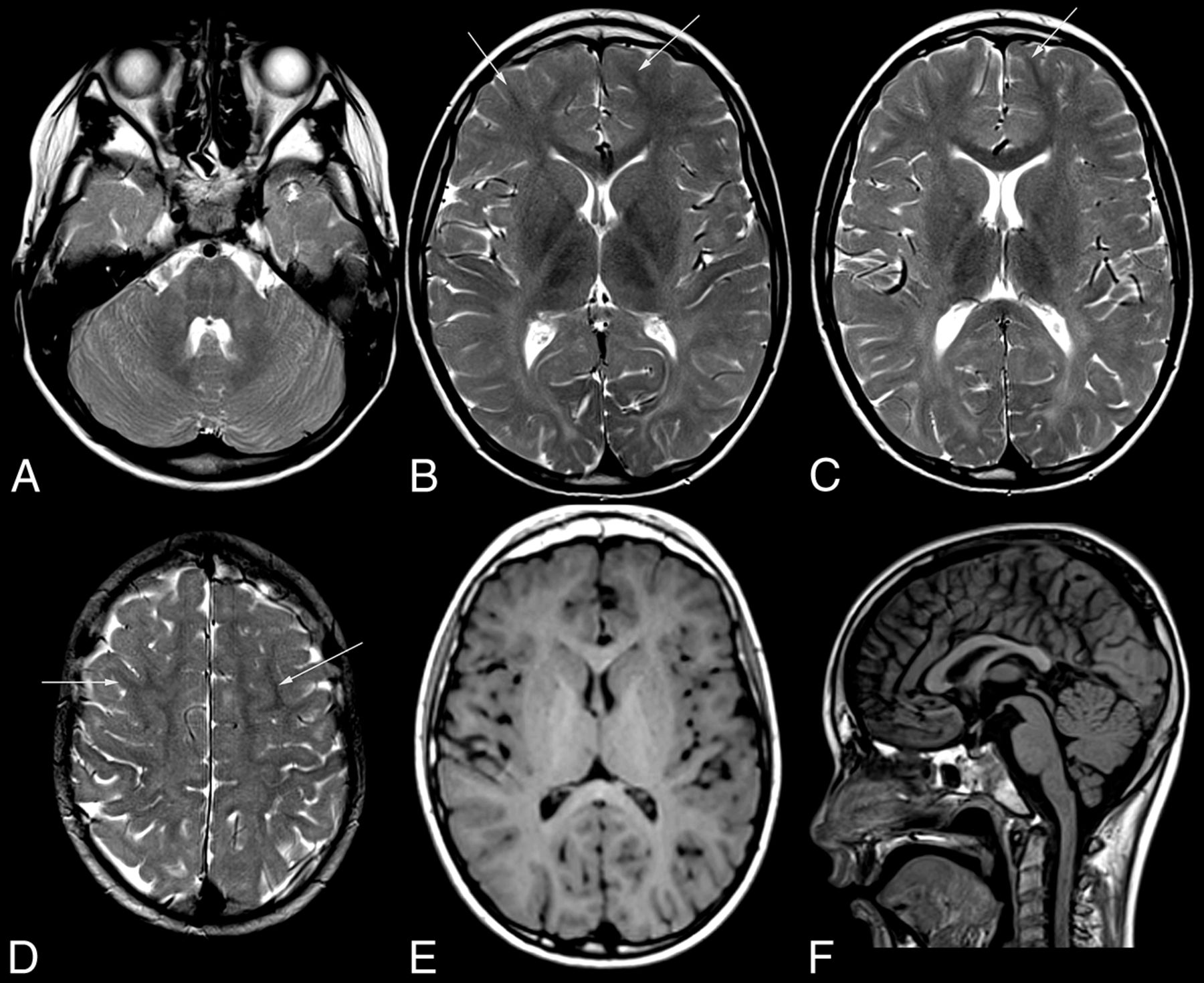
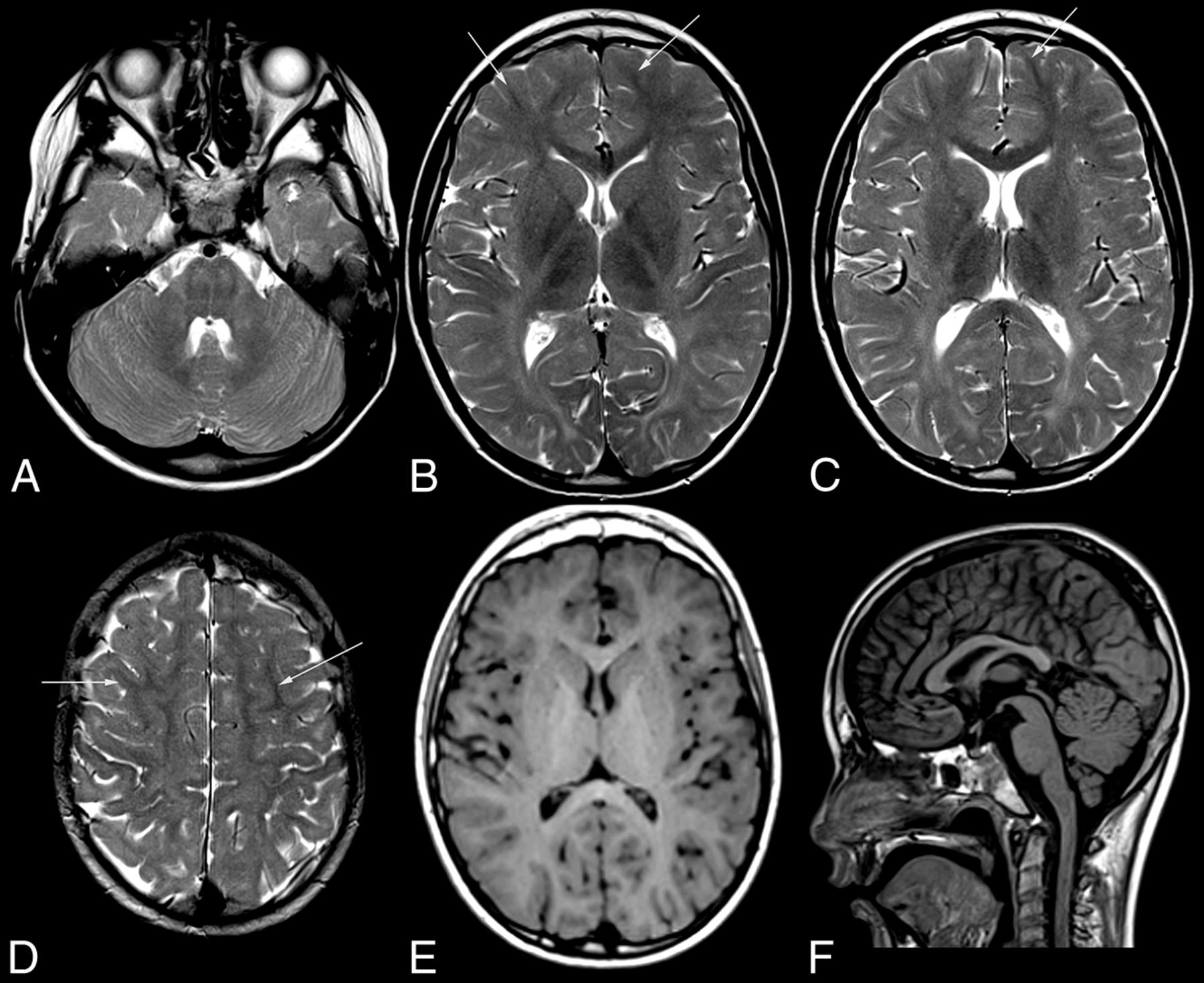
MR imaging pattern of ODDD. Mild, diffuse T2 hyperintensity of the supratentorial white matter (B–D) and normal T1 signal (E) characteristic of hypomyelination in patient 1 with involvement of the posterior limb of the internal capsule (B), lateral splenium (C), and typical brain stem tract involvement at the level of the pons (A). Note T2 hypointensity of the Rolandic cortex compared with subcortical NAWM (arrows) of the medial and superior frontal gyri (B–D) and of the primary visual cortex compared with the anterior limb of the internal capsule, genu, and subcortical frontal NAWM (B and C). The craniocervical junction is normal (F). For an overview of MR imaging changes of all patients see On-line Fig 1.
Infratentorially, all patients had T2 hyperintensity of the pontine pyramidal tract, medial lemniscus, raphe, and middle cerebellar peduncles, as well as a small, triangular T2 hyperintensity in the lateral pons with its base at the anterolateral pons, located medial to the entry zone, and an intraparenchymal course of the trigeminal nerve, consistent with the anterolateral portion of the middle cerebellar peduncle within the pons. T2 hyperintensity resulted in a typical pattern of signal changes at the level of the pons (Fig 2). In the youngest patient, the pontine pyramidal tract only became clearly T2 hyperintense at 7.5 years of age; in patients 3 and 4, the T2 hyperintensity extended into the pyramis. Inconsistently involved infratentorial white matter structures were the inferior cerebellar peduncle, superior cerebellar peduncle, and decussation of superior cerebellar peduncles (n = 2, 1, and 1, respectively; On-line Fig 2).
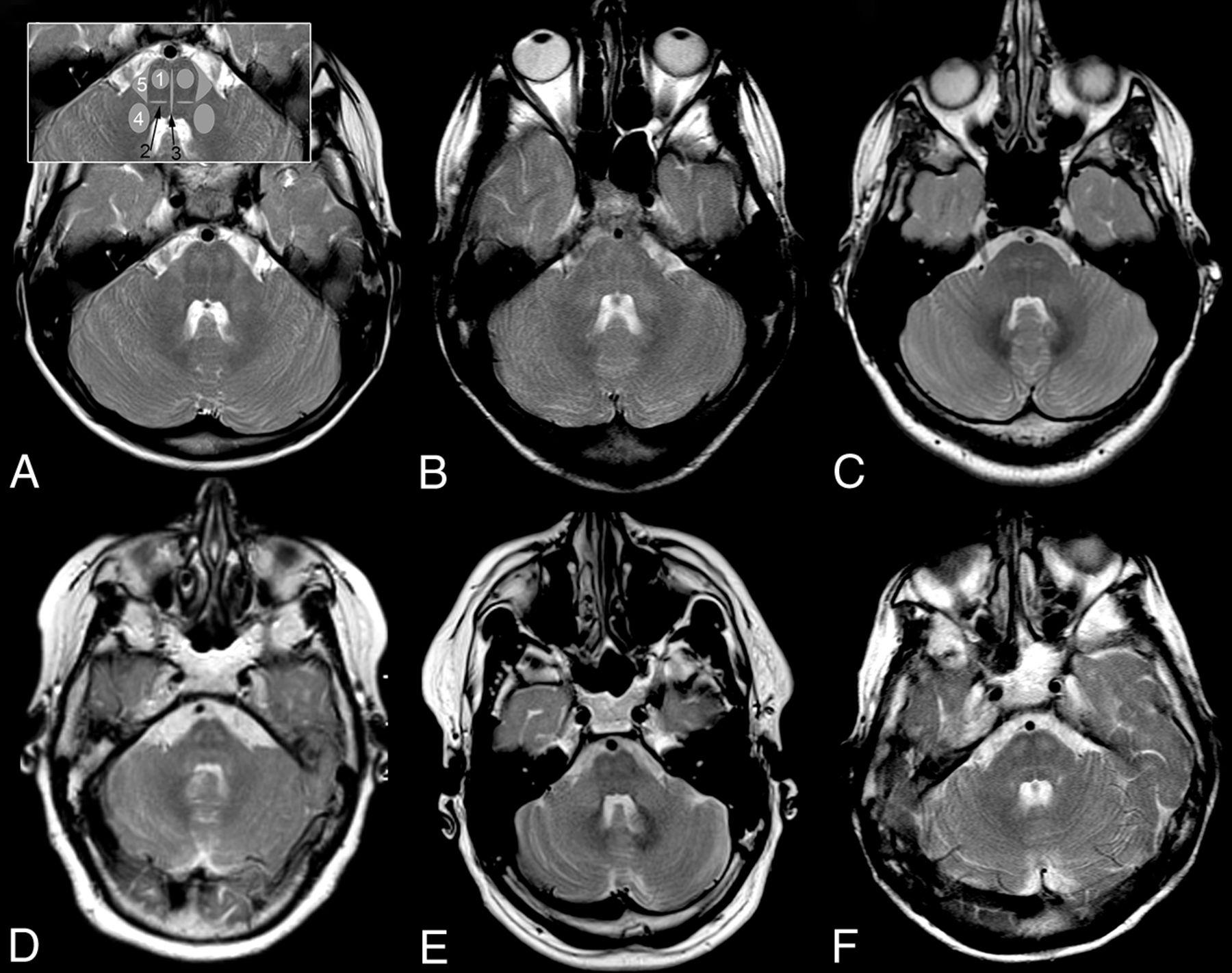
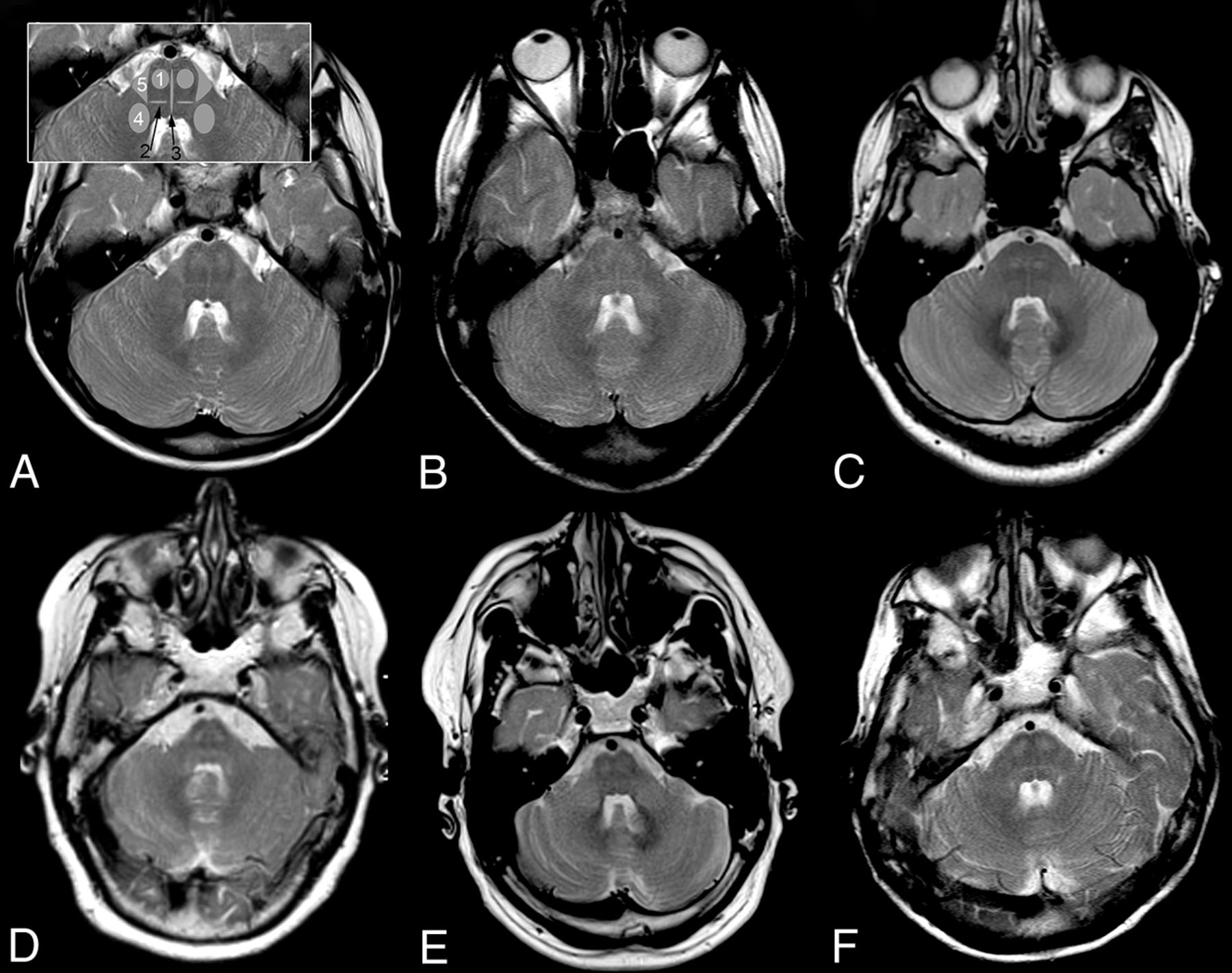
Pattern of brain stem tract involvement. T2WI at the level of the pons demonstrates characteristic involvement of the pyramidal tract (1), lemniscus medialis (2, black arrow), raphe pontis (3, black arrow), middle cerebellar peduncle (4), and the triangular lateral pontine T2 hyperintensity consistent with the anterior course of the pontocerebellar fibers forming the middle cerebellar peduncle (5); schematic representation of tracts is given in inset in A 2, 3 in black. A, Patient 1, B, patient 2, C, patient 3, D, patient 4, E, patient 5, F, patient 6. Field strength is 3T in patient 3; otherwise, it is 1.5T. Images are from the last scan from patients with multiple scans.
The Rolandic and primary visual cortices appeared abnormally T2 hypointense, in patient 1 with hypo- instead of hyperintensity to NAWM visible in the same slice (Fig 1B, D) but without corresponding T2* hypointensity in the 4 patients with T2*/SWI. Due to white matter hyperintensity, the deep gray matter generally appeared relatively T2 hypointense. Compared with age-matched controls, however, only the pallidum (patients 2 and 6) and dentate nucleus (patients 3 and 5) were more T2 hypointense in 2 patients each, the pallidum with corresponding T2* hypointensity in both, T1 hyperintensity in patient 2 (On-line Fig 2), and the dentate without corresponding hypointensity in patient 5 with T2*WI.
DWI did not reveal areas of restricted diffusion. Although in some patients, the ADC was slightly increased in affected white matter areas, this was not very prominent. The craniocervical junction was depicted on sagittal images at least once in all patients, without evidence of stenosis or compression of the medulla.
White matter 1H-MR spectroscopy findings were normal in patient 3. In the other 5 patients, Cr was high or high-to-normal; mIns was high, high-to-normal, or normal; NAA was normal or low-to-normal; and Cho was normal, low-to-normal, or low. These findings resulted in low or low-to-normal metabolite ratios of Cho/Cr and NAA/Cr, consistent with hypomyelination (On-line Table 2). Lactate was not detected, and thus was normal.
Discussion
While syndactyly in combination with typical facial features suggests ODDD, oligosymptomatic patients are common and – in particular if syndactyly is absent – difficult to recognize. Usually only patients with neurologic presentation, occurring in approximately 30%, will be referred for cranial MR imaging. In individual patients with MR imaging previously reported, white matter changes have commonly been observed,2,4⇓–6,13⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓–25 including brain stem involvement in 34⇓–6 and, less commonly, T2 hypointensity of cortical or deep gray matter structures.2,5,6,14⇓–16,22,23 As yet, there are no histopathologic data.
The pattern of brain structures involved in leukodystrophies has been shown to be highly specific and effective for a differential diagnosis using a practical diagnostic algorithm, with differentiation between hypomyelinating and nonhypomyelinating leukodystrophies as the first discriminator.26 Hypomyelination is characterized by moderate T2 hyperintensity, less bright than in nonhypomyelinating leukodystrophies, eg, demyelination or vacuolation. T1 signal depends on the amount of myelin deposited and is often normal or otherwise iso- or mildly hypointense.26,27 On 1H-MR spectroscopy, elevated Cr and mIns in combination with normal or near-normal NAA, low (-normal) NAA/Cr, and low (-normal) Cho are typical.8,9
In all MRIs of our patients, supratentorial white matter changes were characteristic of hypomyelination and accompanied by typical 1H-MR spectroscopy findings in 5 of 6. Three patients imaged during late adolescence and adulthood had foci of more pronounced white matter changes consistent with small areas of secondary degeneration with loss of myelin and astrogliosis. Scarce or absent subcortical NAWM and involvement of the anterior limb of internal capsule only in the 3 oldest patients suggest progression of white matter changes with time as does extension of pyramidal tract hyperintensity from the mesencephalon to the pons during follow-up in the youngest patient.
Hypomyelinating leukodystrophies can be differentiated on the basis of additional, characteristic MR findings, eg, as in hypomyelination with atrophy of basal ganglia and cerebellum (H-ABC) or by the presence of non-neurological features, e.g. congenital cataract in hypomyelination with congenital cataract (HCC), hypodontia and/or delayed puberty in 4H syndrome, or syndactyly in ODDD.27 However, systemic manifestations are not obligatory and therefore may not be present in all patients, as seen in patient 3 who presented with mild spasticity, sensory deficit, and neurogenic bladder but without syndactyly. Even within families, signs and symptoms may vary.1 Notably, all our patients, irrespective of the presence or absence of neurologic manifestation or syndactyly, had a characteristic pattern of brain stem tract involvement in addition to supratentorial hypomyelination, which differed from other hypomyelinating leukodystrophies with brain stem involvement. In Pelizaeus-Merzbacher-like disease, there is prominent T2 hyperintensity of the basis pontis, not always restricted to the pyramidal tract28; in 4H syndrome the medial lemniscus is relatively spared29; in hypomyelination of early myelinating structures30 the medulla oblongata and caudal pons are more extensively involved than in ODDD; moreover, none of these disorders involve the middle cerebellar peduncles. Whereas hypomyelination with brain stem and spinal cord involvement and leg spasticity31 involves the medial lemniscus and pyramidal tract similar to ODDD, the inferior and superior but not middle cerebellar peduncles are T2 hyperintense.
When using prominent brain stem involvement as a starting point of an MR imaging–based differential diagnosis, ODDD must also be differentiated from 3 nonhypomyelinating leukodystrophies: Brain stem involvement is similar in autosomal dominant leukodystrophy related to duplication of the lamin B1 gene,32 but supratentorial white matter changes are more T2 hyperintense, not consistent with hypomyelination, and tend to spare a periventricular rim. T2 hyperintensity of the middle cerebellar peduncles in fragile X premutation carriers is combined with T2 hyperintensity of the deep cerebellar white matter, while the medial lemniscus and pyramidal tract are not affected.33 Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation34 involves the medial lemniscus and pyramidal tract, inferior and superior, but not the middle cerebellar peduncle and again differs by much more prominent supratentorial T2 hyperintensity not consistent with hypomyelination.
It has previously been noted that patients with ODDD imaged in the first decade may have MR imaging changes despite the absence of neurologic symptoms.1 Similarly, all 6 patients had extensive pyramidal tract involvement and involvement of the medial lemniscus on MR imaging, irrespective of the presence of spasticity or impaired sensibility. Compression of the medulla at the craniocervical junction, as initially suggested due to a hyperostotic skull base,3 was not confirmed on MR imaging, either in later reports or in our patients.
Earlier reports also noted calcifications of the basal ganglia, which are a likely correlate of the T2 hypointensity reported in the literature5,6,14⇓–16,23 and of pallidal T2/T2* hypointensity in patients 2 and 6, though we cannot exclude abnormal iron accumulation as previously suggested.6 Consistent with previous reports, the Rolandic and primary visual cortices appeared abnormally T2 hypointense, however without corresponding T2* hypointensity and, therefore, not suggestive of either calcification or abnormal brain iron accumulation.
Conclusions
Brain imaging in 6 patients from 4 families with genetically confirmed ODDD shows a characteristic MR imaging pattern of brain stem tract involvement, consisting of the middle cerebellar peduncle, pyramidal tract, and medial lemniscus in combination with diffuse, supratentorial hypomyelination. Recognition of this pattern should trigger genetic evaluation of undiagnosed patients and thereby shorten the diagnostic process.
REFERENCES
- Received December 7, 2018.
- Accepted after revision March 12, 2019.
- © 2019 by American Journal of Neuroradiology





{kind=link}
{kind=link}