
Abstract
SUMMARY: Retinoblastoma is the most common primary intraocular tumor of childhood. Accurate diagnosis at an early stage is important to maximize patient survival, globe salvage, and visual acuity. Management of retinoblastoma is individualized based on the presenting clinical and imaging features of the tumor, and a multidisciplinary team is required to optimize patient outcomes. The neuroradiologist is a key member of the retinoblastoma care team and should be familiar with characteristic diagnostic and prognostic imaging features of this disease. Furthermore, with the adoption of intra-arterial chemotherapy as a standard of care option for globe salvage therapy in many centers, the interventional neuroradiologist may play an active role in retinoblastoma treatment. In this review, we discuss the clinical presentation of retinoblastoma, ophthalmic imaging modalities, neuroradiology imaging features, and current treatment options.
ABBREVIATIONS:
- IAC
- intra-arterial chemotherapy
- IVC
- IV chemotherapy
- IvitC
- intravitreal chemotherapy
- EBRT
- external-beam radiation therapy
- OA
- ophthalmic artery
Retinoblastoma is the most common primary intraocular malignancy in children. Prompt diagnosis is essential to preserving life, eye, and sight. Neuroradiologists play an important role in diagnosis, staging, and treatment of patients with retinoblastoma. In this review, we aim to educate neuroradiologists regarding retinoblastoma imaging features and basic principles of treatment.
EPIDEMIOLOGY
Retinoblastoma affects 1 in 16,000 births,1 with 8000–10,000 children diagnosed annually. There are no known geographic, racial, or sex predilections. Heritable retinoblastoma is diagnosed at a median age of 12 months and nonheritable disease at 24 months, with 80% diagnosed before 4 years of age.
CLINICAL PRESENTATION AND DIAGNOSIS
Retinoblastoma is diagnosed by physical examination and classified as International Classification of Retinoblastoma groups A through E according to increasing severity. The most common presenting feature is a white pupillary reflex called leukocoria, often recognized first by parents. Strabismus and decreased vision are also common. Patients with advanced disease can present with iris color changes, an enlarged cornea and globe, orbital inflammation, and exophthalmos. Biopsy is avoided because of a risk of tumor seeding, though aqueous biopsy is under investigation.
PATHOPHYSIOLOGY
Retinoblastoma results from a biallelic mutation of the retinoblastoma gene (RB-1) in developing retinal cells, following the 2-hit model of tumor suppressor gene inactivation.2 In the heritable form, seen in up to 50% of patients, a germline mutation is followed by a second acquired somatic mutation. In the nonheritable form, 2 somatic mutations must occur in the same gene in a single cell.
Retinoblastoma originates in the retina and can display endophytic growth into the vitreous chamber; exophytic growth into the subretinal space; or diffuse, infiltrative growth along the retina. Most tumors demonstrate both endophytic and exophytic growth and can cause retinal detachment as well as vitreous and subretinal tumor seeding.
OPHTHALMIC IMAGING
Color Fundus Photography
The first step in diagnosing retinoblastoma is a complete dilated eye examination, often performed under anesthesia. Widefield color fundus, anterior segment, and external photography facilitate serial monitoring.
Ultrasonography
Ocular ultrasonography is a critical diagnostic test with resolution approaching 150 microns using 10-MHz frequency. On B-scan ultrasonography, retinoblastoma is an attenuated, round tumor with focal areas of high internal reflectivity, corresponding to calcification, which create shadowing that persists with low sonography gain.3 Sonography biomicroscopy uses higher frequencies (50–100 MHz) to obtain anterior segment images and detect anterior segment, iris, ciliary body, and anterior chamber angle invasion.4
Fluorescein Angiography
Fluorescein angiography uses IV sodium fluorescein dye to allow direct visualization of blood flow through retinal vasculature. The dye emits green fluorescent light (520–530 nm) after excitation by blue light (465–490 nm). Key imaging features that this technique may highlight include iris neovascularization, dilated tumor-associated retinal vessels, intrinsic tumor vasculature, venous leakage, and microvasculature changes.5 Compared with completely perfused tumors, partially perfused tumors may require fewer cycles of intra-arterial chemotherapy (IAC) to achieve regression.6 Fluorescein angiography can also detect treatment-related ischemic side effects.7
Optical Coherence Tomography
Optical coherence tomography uses low-coherence light to provide a high-resolution cross-sectional image of the retina, showing details as small as 10 microns and permitting detection of clinically invisible tumors.8,9 Newer optical coherence tomography angiography can image retinal vasculature without injection of IV dye and may prove useful for future diagnosis and monitoring.10
Neuroradiology Imaging
High-resolution contrast-enhanced MR imaging is important for optimal assessment of key features that impact treatment decisions in children with retinoblastoma, such as optic nerve invasion, extraocular tumor extension, and intracranial disease. Imaging approaches differ per institution and individual preferences with variations regarding the use of small surface coils or multichannel head coils and imaging at 1.5T or 3T magnet strength. In this regard, the European Retinoblastoma Imaging Collaboration has published imaging guidelines for diagnostic evaluation of retinoblastoma that specify hardware considerations, optimal pulse sequences, and acceptable minimum spatial resolution.11 Carefully performed MR imaging can help confirm retinoblastoma diagnosis, exclude alternative diagnoses, assess tumor extent and stage, and provide long-term monitoring. Poor prognostic features of choroidal invasion and optic nerve extension cannot be reliably detected on clinical examination, so MR imaging detection of extraocular disease and intracranial tumor is crucial. MR imaging can also provide intraocular tumor detail in patients with tumor-related media opacity and can detect congenital brain anomalies in patients with retinoblastoma and 13q-deletion syndrome.12
On imaging, retinoblastoma is typically irregular in contour. On CT, retinoblastoma has intermediate attenuation with variable calcification and mild to moderate enhancement. On MR imaging, tumor is slightly hyperintense relative to vitreous on T1-weighted images, is dark on T2-weighted images, shows reduced diffusivity on diffusion-weighted images, and demonstrates heterogeneous enhancement with gadolinium. Tumor calcification may be evident as signal voids on susceptibility-weighted sequences.
Calcification is a key diagnostic feature present in approximately 95% of cases. Although CT has been used historically to detect tumor calcification, ultrasonography has good sensitivity, and because of potential increased risk for second cancers with ionizing radiation exposure, especially in germline retinoblastoma, sonography for detection of calcification has replaced CT, and the role of CT in the evaluation of retinoblastoma is now considered obsolete.13⇓⇓-16
IMAGING APPROACH TO DISEASE
Intraocular Tumor
The smallest intraocular retinoblastomas are well-circumscribed and confined to the retina. These are typically best visualized on heavily T2-weighted 3D images, such as 3D CISS/FIESTA (Fig 1A). Larger endophytic tumors are associated with vitreous seeding and exophytic tumors with subretinal seeding. Diffusely infiltrating retinoblastoma is uncommon, accounts for approximately 2% of cases, presents at an older mean age of 6 years with inflammatory features, and demonstrates linear growth without a discrete mass or calcification.17,18

A 21-month-old boy with right group B and left group E retinoblastoma. Coronal reformat of 3D FIESTA (A) shows a hypointense calcified mass in the left globe with serous retinal detachment. Two of 3 small retinoblastoma tumors in the right globe are shown (black solid arrows). Axial post-gadolinium T1-weighted image with fat saturation (B) shows enhancing tumor abutting the normally enhancing choroid (white arrow) making focal tumor invasion challenging to assess. Tiny foci of enhancement surrounding the left optic nerve cuff represent normal small vessels (black and white arrow). The left globe was larger than the right consistent with buphthalmos. Gross pathology specimen of the enucleated left eye (C) shows the posteriorly located intraocular neoplasm (arrow). Histopathologic examination showed moderately differentiated retinoblastoma without choroidal invasion and with tumor involving the optic nerve head and lamina cribrosa, but without postlaminar tumor.
Anterior Chamber Involvement
Tumor extension into the anterior chamber is uncommon, may be evident by enhancement on MR imaging, and is associated with iris neovascularization, hyperemia, and inflammation.19,20 Anterior chamber, ciliary body, or iris invasion may necessitate enucleation.21,22
Vitreous Seeding
Vitreous involvement can range from small conglomerates of tumor cells to scattered, discrete tumor foci to diffuse involvement. High-resolution MR imaging can identify vitreous seeding as small foci of T2 signal shortening within the vitreous chamber, but clinical examination is more sensitive with a potential role for optical coherence tomography monitoring.23,24
Vitreous Hemorrhage
Vitreous hemorrhage complicates tumor assessment in children with retinoblastoma, obscuring tumor margins on clinical and MR imaging examination, and is present in fewer than 3% of cases.
Subretinal Tumor Invasion
Retinoblastoma that proliferates within the subretinal space between the retina and the choroid is termed subretinal tumor seeding. Subretinal tumor can be complicated by secondary serous retinal detachment and less frequently retinal hemorrhage (Figs 2 and 3).25

Group E retinoblastoma managed with IAC and IvitC. A 2-year-old boy presented with leukocoria in the right eye (A). Ultrasonography (B) revealed a large, round, hyperechoic retinal mass with intrinsic calcifications seen as foci of high reflectivity and serous retinal detachment (white arrow). After 3 cycles of IAC and IvitC using melphalan and topotecan, a follow-up sonography image (C) shows regression of tumor to a calcified hyperechoic scar with persistent serous retinal detachment (black arrow).
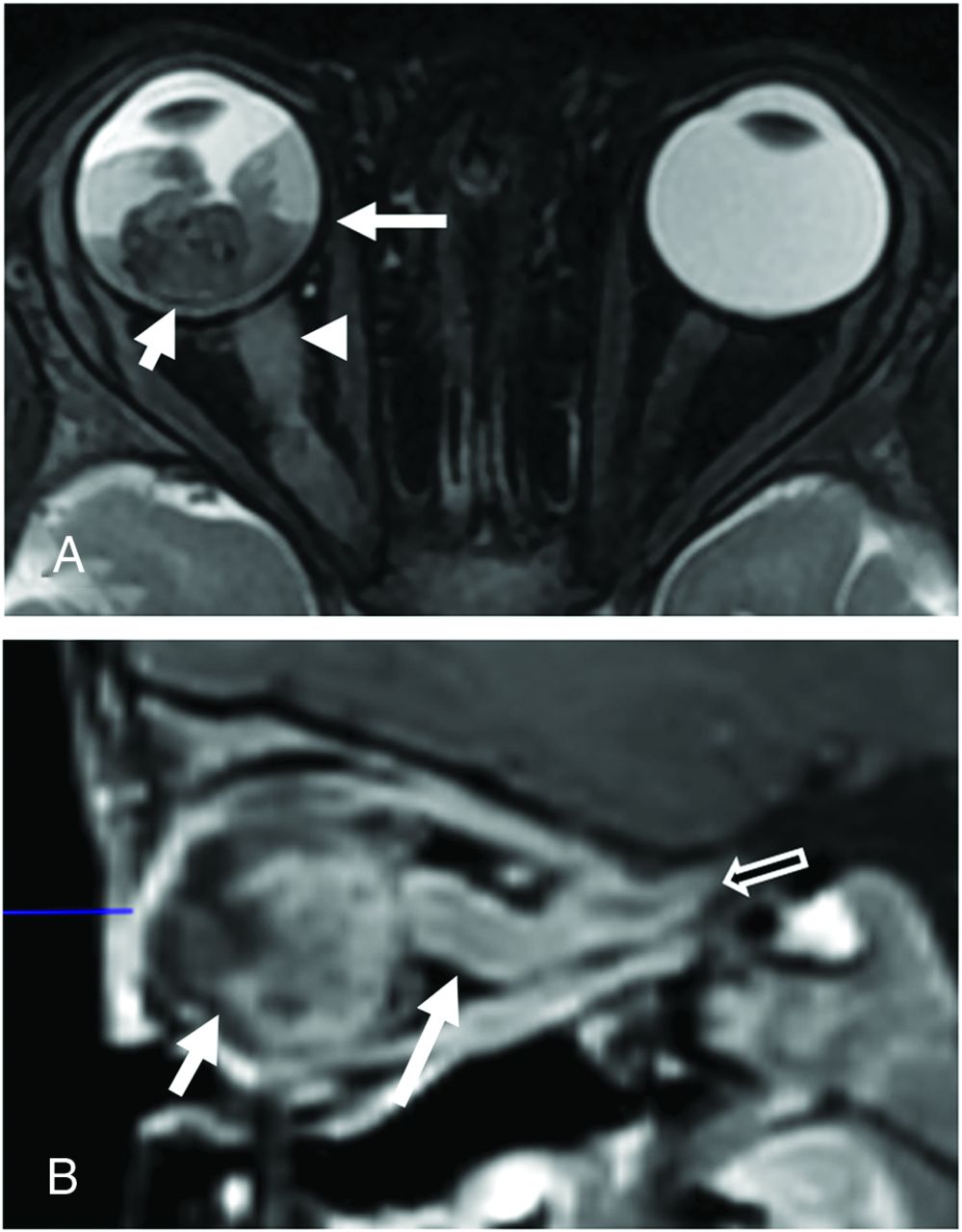
A 21-month-old boy with group E retinoblastoma of the right eye. Axial fat-saturated T2-weighted image (A) shows a large hypointense intraocular tumor (short white arrow) in the right globe with retinal detachment and hemorrhage with a fluid–fluid level (long white arrow). Abnormal thickening of the right optic nerve (arrowhead) is consistent with optic nerve invasion. Sagittal postgadolinium T1-weighted image with fat saturation (B) demonstrates enhancing tumor contacting the lens (short arrow) and tumor invading the optic nerve and extending posteriorly along the optic nerve sheath (long arrow) to the level of the optic canal (open arrow). Histopathologic examination showed poorly differentiated retinoblastoma with choroidal and extensive postlaminar optic nerve invasion.
Choroidal Invasion
Exophytic growth, breaching the Bruch membrane, can result in tumor invasion of the choroid between the retina and sclera. On MR imaging, the choroid normally appears as a linear, uniformly enhancing layer deep to the hypointense sclera. Choroidal invasion is suggested by a focal region of choroidal thickening, either enhancing or nonenhancing, or a focal region of choroidal thinning with decreased enhancement (Fig 1B).25⇓⇓-28 Subtle choroidal invasion remains challenging to discern when enhancing tumor abuts the normally enhancing choroid with accuracies estimated at 57%–72%.25,29 Massive choroidal invasion ≥3 mm is associated with increased metastatic risk, warranting enucleation and adjuvant systemic chemotherapy.30,31
Additional Intraocular Complications
Additional tumor-related complications include a subluxed or dislocated lens, an enlarged globe from secondary glaucoma (Fig 1B), a shallow anterior chamber with increased intraocular pressure, intraocular hemorrhage, and rarely phthisis bulbi.32 Advanced retinoblastoma can cause exophthalmos and periorbital soft tissue swelling from tumor necrosis that can mimic periorbital cellulitis.33
Extraocular Retinoblastoma
Extraocular tumor can manifest by extension of tumor through the sclera into the retrobulbar fat and by growth along the optic nerve and meningeal sheath (Figs 1, 3, and 4).34
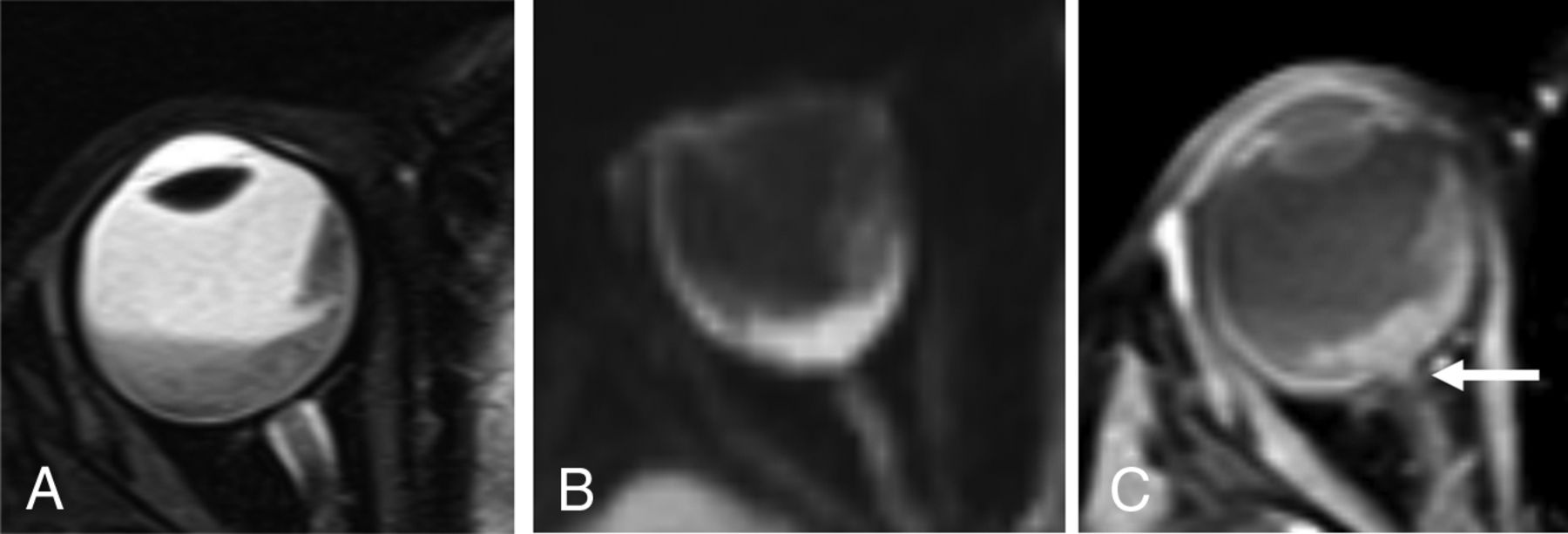
A 2-year-old girl with group E retinoblastoma. Axial fat-saturated T2-weighted image (A) shows a hypointense intraocular tumor centered within the posterior right globe with retinal detachment. Axial trace DWI shows reduced diffusivity of the tumor (B). Axial postgadolinium T1-weighted image with fat saturation (C) shows enhancement of the distal optic nerve in continuity with tumor over 1.2 mm (arrow) and enhancing tumor abutting the enhancing choroid, limiting the assessment for focal choroidal invasion. Histopathologic examination showed poorly differentiated retinoblastoma (grade IV of IV) largely confined to the posterior eye with postlaminar optic nerve invasion and no choroidal invasion.
Scleral and Extrascleral Involvement
Tumor infiltration of the sclera may be apparent on MR imaging by interruption of its normally thin, linear hypointense appearance on both T1-weighted and T2-weighted images and by extrascleral extension of tumor into the retro-ocular fat. The normal discontinuity of the sclera where the optic nerve inserts into the globe should not be mistaken for pathology, though tumor can extend into the optic nerve through the lamina cribrosa at this site. Scleral invasion and extraocular tumor extension are rare in developed countries and signify a poor prognosis and increased likelihood of metastasis.35 Occasionally, enhancement of retrobulbar fat without a tumor mass is observed, which can represent peritumoral inflammation rather than extraocular tumor extension.
Optic Nerve Involvement
Assessment of the optic nerve is essential for accurate staging. Prelaminar and intralaminar invasion of the optic nerve are not considered high-risk features.34 Postlaminar optic nerve invasion occurs in approximately 8% of patients36 and typically requires enucleation followed by systemic chemotherapy because of increased risk for metastatic disease and mortality.28,34,37,38 On MR imaging, the location of the lamina can be estimated at the junction between the optic disc and the midpoint between the enhancing choroid and the low intensity sclera. A definitive diagnosis of postlaminar optic nerve invasion is based on histopathology, which may not be available if eye-preserving treatment is considered. Thickening and enhancement of the postlaminar optic nerve on MR imaging usually indicates tumor extension (Fig 4). However, more subtle abnormalities at the globe–nerve junction can be challenging to accurately classify. Enhancement of the optic nerve at the globe–nerve junction can represent postlaminar tumor, pre- or intralaminar tumor with a posteriorly displaced lamina cribrosa, central retinal vessels, or inflammation (Fig 1).20,36,39⇓⇓⇓-43 Postlaminar optic nerve invasion can be diagnosed based on visual assessment of optic nerve enhancement and tumor size with sensitivity estimated at 59% and specificity estimated at 94%.39,44,45 Lower accuracy may be related to differences in the level of suspicion, imaging technique, and the experience of the interpreting radiologist.46⇓-48 The most reliable direct MR imaging criteria to rule out advanced optic nerve invasion are normal optic nerve size, normal optic nerve signal on T2-weighted images, and optic nerve enhancement of ≤3 mm on postcontrast imaging.36,39 Normal enhancement of the central retinal vessels in the optic nerve and “double-dot” enhancement, seen as punctate foci of enhancement straddling the central retinal vessels at the globe–nerve junction, should not be mistaken for tumor.39 In addition, increased intraocular pressure can cause posterior bulging of the lamina cribrosa that can simulate optic nerve invasion and result in a false-positive interpretation.36,41 MR imaging remains limited in its ability to detect early stage optic nerve invasion with pathology remaining the criterion standard for assessing high-risk features.28,39
TREATMENT MODALITIES
Retinoblastoma treatment is individualized depending on International Classification of Retinoblastoma group staging, laterality of involvement, germline testing results, institutional resources, and psychosocial factors.49 Current treatment modalities include IV chemotherapy (IVC), IAC, intravitreal chemotherapy (IvitC), intracameral chemotherapy, consolidation therapies (cryotherapy and transpupillary thermotherapy), radiation-based therapies (external-beam radiation therapy [EBRT] and plaque radiation therapy), and enucleation.
Intravenous Chemotherapy
Systemic IVC, introduced in the 1990s, remains a key treatment tool, especially for bilateral and germline disease. The most frequently used regimen consists of vincristine, etoposide, and carboplatin (VEC), administered through a central or peripheral catheter monthly for 6 cycles.50 Tumor control is best for less advanced disease.51 Systemic chemotherapy may provide protection against second cancers, metastasis, and pineal embryonal tumor in patients with germline disease.52,53 Intensified chemotherapy regimens are used to treat high-risk disease after enucleation. Neoadjuvant chemotherapy can be used in advanced disease with choroidal or postlaminar optic nerve invasion before enucleation and may prevent exenteration in orbital retinoblastoma.54,55
Intra-arterial Chemotherapy
Introduced in 2004, IAC is now a standard of care first-line therapy, especially for advanced, unilateral tumors, and carries a lower risk of side effects such as ototoxicity and neurotoxicity associated with systemic chemotherapy.56⇓⇓-59 Melphalan, topotecan, and carboplatin are the most commonly used drugs, administered as combinations of 1, 2, or 3 agents depending on tumor severity.50 Treatment is usually administered monthly for 3 or 4 cycles. IAC can be used as a primary or secondary treatment and has higher success rates for globe salvage in advanced tumors compared with IVC (Fig 5).49,60

Group D retinoblastoma managed with IAC and IvitC. A 7-year-old boy presented with a large, white inferiorly located orbital tumor shown in the color fundus photograph (A). Ultrasonography revealed a round, hyperechoic retinal mass (B). After 4 cycles of IAC and IvitC using melphalan and topotecan, the tumor regressed to a small, calcified scar seen on fundoscopy (C) and confirmed by ultrasonography (D) (white arrow).
An experienced interventional neurosurgeon or neuroradiologist is re-quired to perform this procedure and this approach achieves superselective delivery of 10 times the chemotherapy dose to the eye compared with IVC.61 The target artery into which the chemotherapy is administered is the central retinal artery, a branch of the ophthalmic artery (OA). The central retinal artery is too small to visualize on conventional angiography, so the posterior ciliary arteries, which cause the choroid blush, are often used as the surrogate (Fig 6). The OA is the first intradural branch of the internal carotid artery and it is the dominant arterial supply to the retina in more than 98% of adults. However, during fetal life, there are robust connections between the OA and branches of the middle meningeal artery. These connections persist into early childhood, and compared with adults, there may be a higher prevalence of dual supply to the retina from both the middle meningeal artery (ie, the artery of the superior orbital fissure) and the OA (Fig 6).62 Preoperative MRA can detect anatomic variations.

Competitive flow to the choroid from the OA and middle meningeal artery (MMA). A lateral angiographic image provides reference for OA supply to the globe (A); the posterior ciliary artery is the vascular supply to the choroid and produces the choroid blush. This is the imaging surrogate for the origin of the central retinal artery. Right ICA injection (B) shows the normal OA origin (black arrow) and expected choroid blush (white arrow). Right ECA injection (C) shows the artery of the superior orbital fissure (Art SOF; black and white arrows), which anastomoses with distal branches of the OA and then fills the OA in a retrograde fashion (black arrows) indicating competitive flow between the MMA and OA for supply to the retina. MR angiogram (D) shows supply to the orbit through the artery of the superior orbital fissure (white arrow), a branch of the MMA (white and black arrow).
Regarding technical aspects, IAC delivery is a relatively straightforward procedure. At our center, we typically place a 4F Berenstein catheter (Boston Scientific) in the femoral artery and advance it to the carotid artery on the side of the lesion. Internal and external carotid artery injections are performed to identify the source of the choroid blush. After this, a microcatheter (Headway Duo, MicroVention; or Magic, Balt; 1.2F) is advanced in a flow-directed technique (ie, wireless) to the ostium of the OA, and after confirmation of positioning, the chemotherapeutic agents are administered over the course of 1 hour.
Complications can occur. Catheterization and contrast injection into the OA can trigger an oculocardiac reflex and severe bradycardia. Ocular side effects can include transient periorbital edema, cutaneous hyperemia, madarosis, blepharoptosis, scalp hair loss, and extraocular motility abnormalities.63⇓-65 More lasting vision-threatening ophthalmic vascular events include vitreous hemorrhage, retinal and choroidal ischemia with resulting atrophy, branch or central retinal artery occlusion, and OA spasm or occlusion.7,60,63,66⇓-68 Retinal detachment occurs in up to 25% of treated eyes presumably because of rapid tumor regression. The rate of serious ophthalmic vascular events that result in chorioretinal atrophy and vision loss has decreased over time and correlates with increased experience of the treating physician.68 Risks are similar when IAC is used as primary or secondary therapy.69 Complications related to chemotherapy administration include neutropenia, fever, and intraoperative bronchospasm.
Intraocular Chemotherapy
IvitC is used in combination with IAC to treat vitreous seeding. The most common drugs are melphalan and topotecan, used alone or in combination. Treatment is administered every 2–4 weeks and tailored to tumor response. Globe salvage for group E eyes has improved to >50% by combining IAC and IvitC (Fig 5).70,71 Small chemotherapy doses can be delivered to the anterior chamber (intracameral chemotherapy) for aqueous seeding.72
Focal Therapies
Focal therapies may be the primary treatment for small retinoblastomas but are typically used for consolidation in combination with IVC.73 Whereas cryotherapy with a triple-freeze–thaw technique is used for peripheral tumors, transpupillary thermotherapy with a diode laser is typically used for posteriorly located tumors that cannot be reached with the cryotherapy probe.
External-Beam Radiation Therapy
EBRT is generally avoided because of associated side effects, the most serious of which is increased risk for second primary malignancies, especially in patients with germline retinoblastoma.16,74,75 EBRT maintains a role in treating extraocular tumor extension, orbital recurrence, and a positive optic nerve margin following enucleation.76
Plaque Radiation Therapy
Plaque radiation therapy, also known as brachytherapy, delivers localized, low-dose radiation, often as secondary treatment for small- to medium-sized tumors that are chemoresistant or for localized recurrence.77 Unlike EBRT, plaque radiation therapy is not associated with increased risk for second cancers.78,79
Enucleation
Enucleation is sometimes necessary to protect the life of patients with advanced disease. Enucleation is typically reserved for advanced group E tumors, especially if there is poor visualization of the fundus or concern for optic nerve or extraocular extension.21,80⇓⇓-83 Tumors that have failed other therapies may also require enucleation. If there are high-risk features on histopathology of the enucleated globe, additional systemic chemotherapy is required for prevention of metastatic disease.84
Advanced Disease
Trilateral Retinoblastoma.
Trilateral retinoblastoma refers to the presence of intracranial tumor histologically similar to retinoblastoma in the pineal gland or suprasellar cistern (supratentorial CNS embryonal tumor) in addition to bilateral ocular involvement.85 This occurs in 10%–15% of patients with heritable retinoblastoma.86 The presence of CNS tumor in both midline locations is sometimes referred to as quadrilateral retinoblastoma (Fig 7). The incidence of trilateral retinoblastoma is 3.2%. These intracranial tumors can be synchronous with intraocular retinoblastoma at diagnosis but are more frequently found on surveillance MR imaging. The lead time from asymptomatic to symptomatic pineal trilateral tumor is approximately 1 year. The prognosis for trilateral retinoblastoma is poor.87⇓-89

A 16-month-old boy presented with extensive metastatic disseminated quadrilateral retinoblastoma. Coronal T2-weighted image from the initial MR imaging (A) showed hypointense retinal tumors in both globes (black arrows). Sagittal postgadolinium T1-weighted image from the initial MR imaging (B) showed a large enhancing mass occupying the sella, suprasellar cistern, and third ventricle, with separate tumors involving the pineal gland and extending along the planum sphenoidale. Sagittal postgadolinium T1-weighted image from an MR imaging 5 weeks later (C) shows tumor growth, obstructive hydrocephalus, and leptomeningeal dissemination of tumor within the fourth ventricle and along the brain stem and upper cervical cord (white arrows).
Scrutiny of the pineal gland at presentation and on surveillance MR imaging is important for detecting developing pineal region tumor. It is important to not mistake pineal cysts for tumor; pineal cysts are a common incidental finding in the general pediatric population, particularly at 3T. Incidental pineal cysts may grow, have proteinaceous content, and may show enhancement of the cyst wall with mild central enhancement.90,91 Embryonal tumors of the pineal gland, on the other hand, have imaging characteristics similar to retinoblastoma and are predominantly solid and dark on T2-weighted images, enhance, and show reduced diffusivity (Fig 7).
CNS Dissemination and Metastatic Disease.
Tumor spread into the intracranial space is uncommon and carries a poor prognosis. Retinoblastoma can spread through direct extension via the optic nerve and its meningeal sheath into the intracranial compartment and can metastasize systemically via hematogenous or lymphatic routes (Fig 7).28,34 The most commonly affected lymph nodes are in the preauricular and cervical regions. Systemic metastases are most frequently observed in the lung, liver, bone, and brain.
Tumor Surveillance and Treatment-Related Findings
Posttreatment imaging includes tumor surveillance to identify local or intracranial recurrence, new intracranial or spinal CSF seeding, and development of trilateral or quadrilateral retinoblastoma. The contralateral globe must be monitored to identify metachronous tumor. Treatment-related complications include cataract, vitreous hemorrhage from radiation retinopathy, enophthalmos, phthisis bulbi, myositis, nonspecific inflammatory changes, enhancement of orbital fat and optic nerve, and orbital and midface hypoplasia from EBRT.92⇓-94 Secondary tumors are seen in heritable forms of retinoblastoma, especially after radiation. Patients with germline disease are at increased risk for osteosarcoma and soft tissue sarcoma, melanoma, and malignancies of the brain and nasal cavities. The cumulative incidence for new cancer at 50 years after retinoblastoma diagnosis is 36% for those with hereditary retinoblastoma and 5.7% for those with nonhereditary retinoblastoma.75
CONCLUSIONS
Retinoblastoma is a complex disease that requires careful work-up for accurate diagnosis and an expert, multidisciplinary team for appropriate treatment, optimized vision-sparing survival, and long-term surveillance. The neuroradiologist is a critical part of the retinoblastoma care team and should be familiar with key diagnostic and prognostic features of this disease.
Indicates open access to non-subscribers at www.ajnr.org
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- Received June 9, 2020.
- Accepted after revision October 14, 2020.
- © 2021 by American Journal of Neuroradiology




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}