
Abstract
SUMMARY: Autoimmune encephalitis is a relatively new category of immune-mediated disease involving the central nervous system that demonstrates a widely variable spectrum of clinical presentations, ranging from the relatively mild or insidious onset of cognitive impairment to more complex forms of encephalopathy with refractory seizure. Due to its diverse clinical features, which can mimic a variety of other pathologic processes, autoimmune encephalitis presents a diagnostic challenge to clinicians. Imaging findings in patients with these disorders can also be quite variable, but recognizing characteristic findings within limbic structures suggestive of autoimmune encephalitis can be a key step in alerting clinicians to the potential diagnosis and ensuring a prompt and appropriate clinical work-up. In this article, we review antibody-mediated encephalitis and its various subtypes with a specific emphasis on the role of neuroimaging in the diagnostic work-up.
ABBREVIATIONS:
- NMDA
- N-methyl D-aspartate
- NMDAr
- N-methyl D-aspartate receptor
- VGKC
- voltage-gated potassium channel
Autoimmune encephalitis is an important cause of new-onset altered mental status, the scope of which has only recently begun to be recognized in the medical literature.1⇓–3 Despite this increased recognition, it has yet to become an established diagnostic consideration outside of large tertiary referral centers.1⇓⇓⇓–5 The term “autoimmune encephalitis” generally refers to a family of closely related disease processes that share overlapping clinical features and neuroimaging findings but are ultimately differentiated by the specific antibody subtypes driving the underlying immune-mediated attack on different CNS structures.6⇓–8 This antibody-mediated attack on neuronal structures results in a localized inflammatory response. Thus, the clinical and imaging manifestations are dictated by the specific location of the underlying immune response within the nervous system, which leads to substantial variability. While limbic dysfunction is the single most consistent finding in autoimmune encephalitis, varying degrees of involvement are seen within the neocortex, striatum, hindbrain, spine, and peripheral nervous system based on the unique antibody profile.3,9⇓⇓–12 In addition, certain antibody subtypes consistently lack imaging manifestations, while others characteristically demonstrate prominent “extralimbic” involvement.3,7,13⇓–15
Although it was initially thought to be relatively rare, there is growing consensus that autoimmune encephalitis is responsible for a subset of altered mental status previously considered idiopathic.3⇓–5 Despite its growing recognition as a rare cause of altered mental status, autoimmune encephalitis remains a diagnosis of exclusion with more common causes often identified during the standard diagnostic evaluation.16,17 However, more complex presentations of altered mental status may display atypical features without a clear etiology identified after routine testing.16,17 In these situations, recognition of potential cases of autoimmune encephalitis by the radiologist can be the first step to optimizing clinical outcomes through ensuring that a prompt and appropriate clinical work-up is performed, including the use of specialized serum/CSF antibody panels, with the ultimate goal of establishing an effective treatment regimen before the onset of devastating complications.3,5,8,18
The purpose of this article is to discuss the subset of immune-mediated CNS conditions with features of autoimmune encephalitis (ie, antibody-mediated inflammation of the brain), provide a framework for radiologists to understand the relevant neuroimmunology, review the major antibody subtypes, and describe the constellation of clinical and imaging features that are most suggestive of this diagnosis.
Pathophysiology: Models for Disease Classification
Antibody-mediated CNS disorders can be classified into 2 broad categories, paraneoplastic or nonparaneoplastic, based on the presence or absence of an underlying malignancy, respectively.18⇓–20 Paraneoplastic syndromes affecting the CNS are generally thought to develop in cancer when antigens shared by tumor cells and native nonneoplastic neuronal cells result in an antibody-mediated attack on previously immune-privileged neuronal structures.2,6⇓–8,19 Initially thought to occur in <1% of patients with cancer, more recent data suggest that the true incidence is likely much higher.3⇓⇓–6 Paraneoplastic syndromes are most often seen in small-cell lung cancer but can also be seen in a variety of other cancers as well, such as neuroblastoma, germ cell tumor of the testis, breast cancer, Hodgkin lymphoma, thymoma, and immature ovarian teratomas.19⇓⇓–22
Regardless of the etiology and antibody profile, there is a clear predilection in autoimmune encephalitis for antigens within the limbic system (Figs 1 and 2).3,10,23,24 Paraneoplastic limbic encephalitis, a specific paraneoplastic syndrome affecting the temporal lobe and limbic structures, was first described by the British neuropathologists Corsellis et al25 in 1968 after identifying postinflammatory changes in the mesial temporal lobes of patients with progressive memory loss after being diagnosed with lung cancer. Kohler et al26 later correlated these inflammatory changes with T2-weighted hyperintense signal changes on MR imaging of the brain. These characteristic neuroimaging findings were later validated by a larger study of 50 patients with paraneoplastic limbic encephalitis across different antibody profiles that found that 39 of 50 patients (79%) had similar T2-FLAIR hyperintense signal changes in their temporal lobes and limbic structures.19 This study, conducted by Gultekin et al19 in 2000, proposed the first diagnostic criteria for paraneoplastic limbic encephalitis, which included the following: 1) short-term memory loss, seizures, or psychiatric symptoms; 2) <4 years between symptom onset and cancer diagnosis; 3) exclusion of metastases, infection, metabolic, or other causes; and 4) one of the following: inflammatory CSF findings, temporal lobe T2 or FLAIR hyperintensity on MR imaging, or electroencephalogram abnormality in the temporal lobes.19 Tüzün and Dalmau27 subsequently modified these criteria in 2007 to account for the growing subset of nonparaneoplastic forms of autoimmune encephalitis, which also demonstrated prominent limbic involvement.

Anti-Hu encephalitis. A 68-year-old man with chronic obstructive pulmonary disease presented with gradually worsening memory deficits and confusion, with subclinical seizures. MR imaging of the brain demonstrates T2-FLAIR hyperintensity and mild expansion in the right medial temporal lobe (A), right insular cortex (not shown), and left dorsal thalamus (not shown), without restricted diffusion (not shown) or postcontrast enhancement (not shown). FDG-PET of the brain demonstrates a hypermetabolic focus within the right medial temporal lobe lesion (B). PET of the body demonstrates a hypermetabolic focus in the left lung (E), consistent with biopsy-proved small-cell lung cancer. The patient was in remission following treatment with intravenous immunoglobulin infusions, oral steroids, and chemotherapy, but he presented approximately 2.5 years later with worsening memory decline. MR imaging at that time (C and D) shows new T2-FLAIR hyperintensity in the left medial temporal lobe (white arrow) with volume loss within the right medial temporal lobe (white arrowhead). An old right occipital lobe infarct is also incidentally noted.

Graves ophthalmopathy with anti-Hu encephalitis. A 63-year-old woman with severe encephalopathy and diffuse enlargement of the extraocular muscles developed fatal autonomic dysfunction. MR imaging of the brain demonstrates prominent T2-FLAIR abnormalities in the mesial temporal lobes (A), right thalamus (B), right > left insular cortex (B), and posterior right temporal lobe (B), without enhancement (C) and with T2 shinethrough but no restricted diffusion on DWI (D) and the corresponding ADC map (E). There is also diffuse symmetric enlargement of the extraocular muscles, resulting in exophthalmos (F).
In addition to the “paraneoplastic-versus-nonparaneoplastic” categorization, antibody-mediated encephalitides can also be characterized as either group I or group II according to the location of their neuronal antigens (On-line Table), with group I antibodies targeting intracellular antigens and group II antibodies targeting antigens on the cell surface.1,2,6,7,9,27 This distinction is clinically relevant because it has implications for treatment response, association with an underlying malignancy, and overall long-term prognosis.9,20,22,28
Group I Antibodies: Autoimmune Encephalitis with Intracellular Antigens
Group I antibodies target intracellular neuronal antigens, are more closely associated with an underlying malignancy, and use the same cytotoxic T-cell mechanisms when targeting the intracellular neuronal antigens and onconeuronal antigens as part of the immune response to cancer.1,7,9,29 Group I antibodies are also associated with poor clinical outcomes, characterized by a decreased response to immunomodulatory therapy and an increased prevalence of “irreversible” neuronal damage, and often have the additional burden of an underlying malignancy.9,19,21 Compared with group II antibodies, group I antibodies are less specific clinical markers of disease for autoimmune encephalitis and can also be seen in patients with cancer without paraneoplastic syndromes.29,30
Anti-Hu
Anti-Hu (anti-neuronal nuclear antibody 1) encephalitis is the most common paraneoplastic form of autoimmune encephalitis, has a relatively poor prognosis compared with other subtypes, and is associated with small-cell lung cancer in most cases (75%).19,31 Anti-Hu syndrome is a distinct clinical phenotype described in patients with cancer expressing anti-Hu antibodies and has features of paraneoplastic encephalomyelitis, paraneoplastic subacute sensory neuropathy, and paraneoplastic cerebellar degeneration.19,31,32 While anti-Hu encephalitis is not as closely associated with seizures as some of the other major subtypes of autoimmune encephalitis, a subset of patients with anti-Hu encephalitis can present with epilepsia partialis continua, a specific seizure disorder characterized by extended focal motor epileptic seizures prominently involving the face and distal extremities that recur every few seconds/minutes.32,33 MR imaging findings correlate with clinical features and typically include T2-FLAIR hyperintense lesions in the medial temporal lobes with variable involvement of the cerebellum and brain stem (Figs 1 and 2).3,19,21,23
Anti-Ma/Ta
Anti-Ma (Ma1/Ma2/Ma3) encephalitis has a better prognosis than anti-Hu and is strongly associated with testicular tumors in young men and small-cell lung cancer or breast cancer in older patients.7,9,34,35 The association with testicular tumors in young men is so strong that some authors have advocated empiric orchiectomy in refractory cases of severe anti-Ma encephalitis for presumed microscopic neoplastic testicular tumors if certain diagnostic criteria are met and no other etiology is found.36 According to a review of 38 patients with anti-Ma encephalitis, most patients (62%) presented with neurologic symptoms before the identification of their malignancy, which included any combination of limbic, diencephalic, or brain stem dysfunction.13 Notably, only a minority of patients (26%) had classic symptoms of limbic encephalitis, and most patients with brain stem involvement had ophthalmoplegia (92%).13 Abnormal findings on brain MR imaging were common (74%) and often involved classic T2-FLAIR hyperintense lesions in the medial temporal lobes with variable involvement of the thalamus and brain stem.13 Although not classic, nodular postcontrast enhancement that can mimic tumor or infection has also been described.13,35,36
Anti-CV2
Anti-CV2 (collapsin response mediator protein 5) encephalitis is a unique subtype associated with small-cell lung cancer and malignant thymoma that has prominent T2-FLAIR hyperintense lesions in the striatum and clinically resembles choreiform movement disorders.3,37 MR imaging features are also atypical compared with other types of autoimmune encephalitis in that there is less prominent involvement of the medial temporal lobe.3,37 Most important, there is typically no restricted diffusion or T2-FLAIR hyperintense lesions in the striatum, which can help differentiate this condition from prion diseases like Creutzfeldt-Jakob disease.3,16 When one considers this relatively rare diagnosis, it is important to first rule out more common toxometabolic disorders such as hyperammonemia, carbon monoxide poisoning, and hypoglycemia.
Anti-Glutamic Acid Decarboxylase
Glutamic acid decarboxylase (GAD) is an intracellular enzyme that catalyzes the synthesis of γ-aminobutyric acid, the major inhibitory neurotransmitter in the CNS. Anti-glutamic acid decarboxylase antibodies are unique because they are a group I antibody not typically associated with malignancy and are also associated with other nonneoplastic autoimmune conditions such as type 1 diabetes mellitus.9,38 The anti-glutamic acid decarboxylase antibody subtype can cause a form of autoimmune encephalitis with classic temporal lobe lesions on MR imaging with the expected clinical findings of limbic encephalitis plus additional features of stiff person syndrome with early and prominent development of seizures (Fig 3).9,38
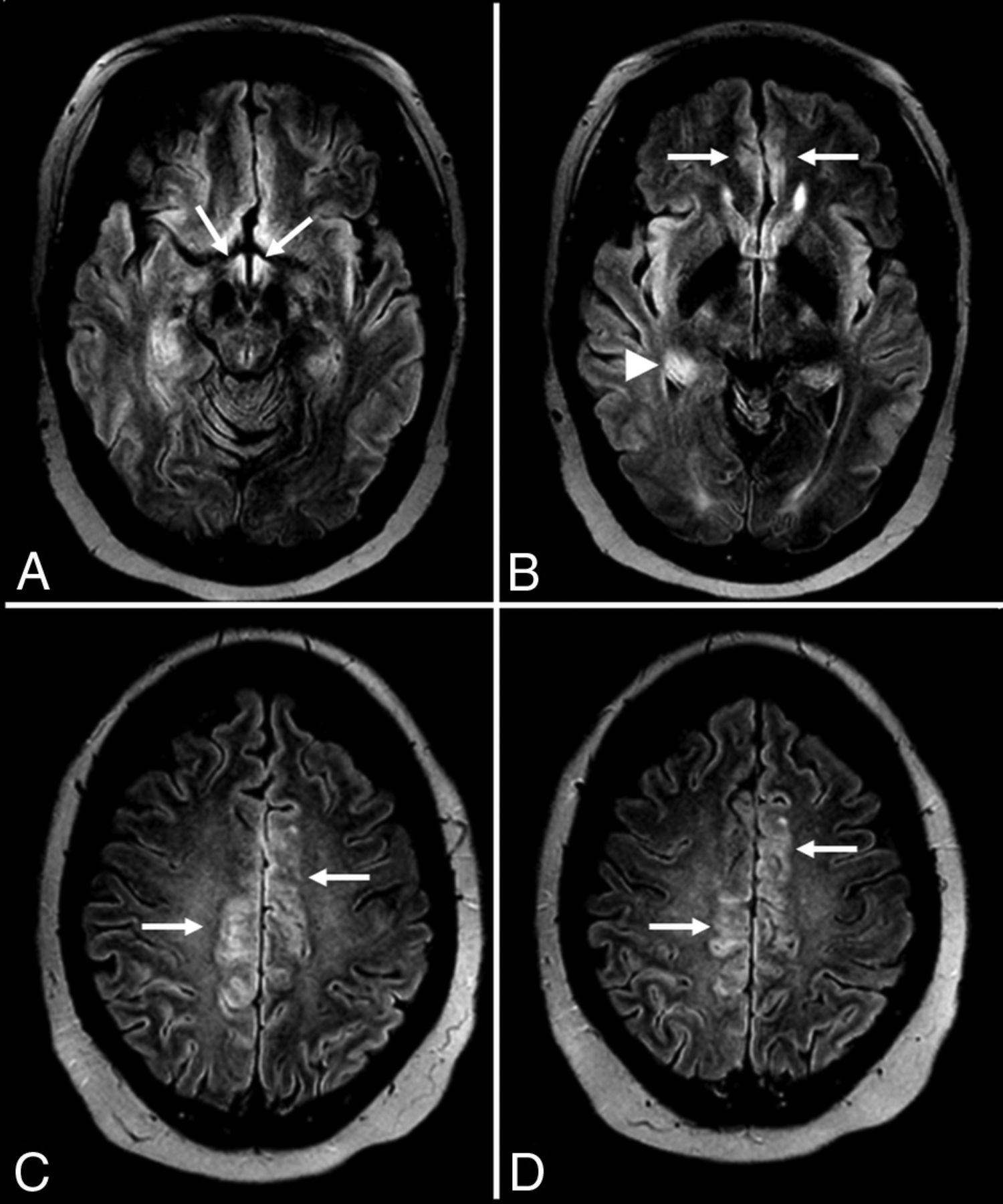
Anti-glutamic acid decarboxylase encephalitis. A 61-year-old woman presented with headaches, mild confusion, and nystagmus without development of psychosis, severe encephalopathy, or seizures. MR imaging of the brain demonstrates T2-FLAIR hyperintensity in the right > left hippocampus (A and B), right > left insular cortex (B), and bilateral cingulate gyrus (C and D) without restricted diffusion (not shown), hemorrhage (not shown), or postcontrast enhancement (not shown).
Additional Type I Antibody Subtypes
Amphiphysin antibodies are most often seen in breast cancer and small-cell lung cancer with associated clinical features of stiff person syndrome, myelopathy, myoclonus, and encephalomyelitis.7,39 Ri (anti-neuronal nuclear antibody 2) antibodies are also most often seen in breast cancer and small-cell lung cancer, with features of brain stem encephalitis and opsoclonus-myoclonus syndrome.7,39 Yo (parietal cell autoantibodies 1) antibodies are most often seen in ovarian cancer and breast cancer, with characteristic features of paraneoplastic cerebellar degeneration, but they can also demonstrate features of autoimmune encephalitis.7,39,40
Group II Antibodies: Autoimmune Encephalitis with Cell-Surface Antigens
Group II antibodies target cell-surface neuronal antigens, are less likely to be associated with an underlying malignancy, and use more “restricted” humoral immune mechanisms of neurotoxicity that typically respond better to early immunomodulatory therapy.9,20,41 Group II antibodies also represent a more specific clinical marker of disease for antibody-mediated encephalitis, with reduction in serum antibody titers following treatment directly associated with improved neurologic outcomes.41,42 Group II antibodies often target synaptic proteins and can result in the downregulation of receptors that leads to altered synaptic transmission associated with epileptiform activity.9,11,15 Patients with non-neoplastic forms of autoimmune encephalitis associated with group II antibodies may have an underlying systemic autoimmune disorder or can develop symptoms following a viral infection or vaccination, but in many cases, no clear etiology is identified.1,4,11,43 The current list of group II antibodies will likely continue to grow on the basis of the number of case reports in the medical literature of “suspected autoimmune encephalitis” or “steroid-responsive limbic encephalitis,” in which a specific antibody or malignancy is not identified but the diagnosis is strongly suggested by a combination of characteristic clinical features, typical neuroimaging findings, good empiric treatment response, and no convincing alternative diagnosis.43⇓–45
N-Methyl D-Aspartate Receptor
N-methyl D-aspartate receptor (NMDAr) encephalitis is one of the most common and best characterized subtypes of autoimmune encephalitis classically seen in young women and children with autoimmunity not associated with cancer (Fig 4).20,28,46 This subtype is mediated by immunoglobulin G antibodies against the GluN1 subunit of the neuronal NMDAr, with inflammatory neuronal dysfunction that is thought to be initially reversible but potentially progresses to permanent neuronal destruction if untreated, due to prolonged inflammation and N-methyl D-aspartate (NMDA)-mediated glutamate excitotoxicity.9,47,48 NMDAr encephalitis has a well-characterized progression of features characterized by an initial viral-like prodrome (fever, malaise, headaches, and anorexia), followed by psychiatric symptoms (anxiety, depression, schizophrenia, and psychosis), which progress to include temporal lobe dysfunction (amnesia and seizures) and ultimately culminate in severe neurologic deficits, including autonomic dysfunction, dystonia/dyskinesia, and profound encephalopathy.3,20,49,50 There are many cautionary reports in the medical literature of young women with NMDAr encephalitis and no significant medical history who present with initial psychiatric symptoms that prompt admission to a psychiatric facility but later require transfer to the intensive care unit after development of the more severe neurologic deficits associated with this condition.18,20,48,51
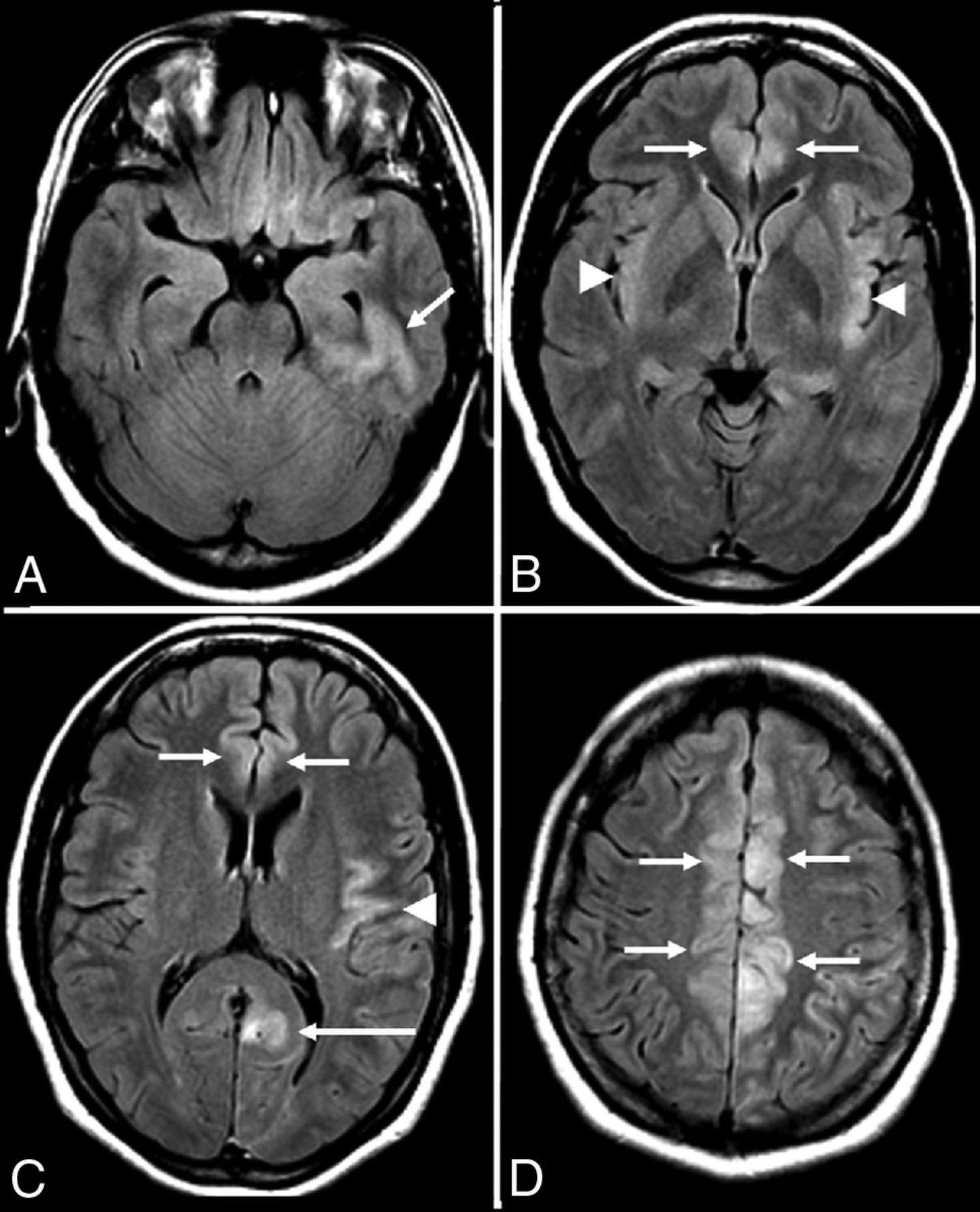
Anti-N-methyl D-aspartate receptor encephalitis. A 32-year-old woman presented with headaches, vertigo, and psychosis with subsequent development of encephalopathy and seizures. MR imaging of the brain performed after the onset of seizures 2 weeks after initial presentation demonstrates T2-FLAIR hyperintensity in the left inferior temporal lobe (A), left > right insular cortex (B and C), and left > right cingulate gyrus (B–D), without restricted diffusion (not shown), hemorrhage (not shown), or postcontrast enhancement (not shown).
With early diagnosis and treatment, patients with NMDAr encephalitis have a relatively good prognosis and can experience a return to their baseline functional status with complete resolution of neuroimaging abnormalities on follow-up examinations.20,41,49 Relapsing forms of nonparaneoplastic NMDAr encephalitis have been reported, and long-term prophylaxis with steroid-sparing agents like rituximab may be required in a subset of cases.42,48,50 A minority of cases of NMDAr encephalitis can be associated with an underlying malignancy, especially in older patients.20,52,53 According to 1 study, 45% of adult women with NMDAr encephalitis had an underlying ovarian teratoma but only 9% of young girls had this finding.53 In women older than 45 years of age, this same study found that 23% of women had an ovarian carcinoma instead of a teratoma.53 This finding highlights the need to screen all patients with autoimmune encephalitis for an underlying malignancy, regardless of the antibody profile, and even to consider the possibility of a contralateral or concurrent tumor with a poor response to treatment despite removal of a tumor.27,49 NMDAr encephalitis is an especially important diagnosis to consider in young patients with limbic encephalitis because the California Encephalitis Project found that the number of young patients in the study with NMDAr encephalitis was greater than those with any single viral etiology.54 Anti-NMDAr antibodies have even been found in patients with herpes simplex virus encephalitis55 and Rasmussen encephalitis,56 which can further complicate the diagnostic work-up.
One unique feature of the NMDAr encephalitis subtype is that it is unlikely to have associated neuroimaging abnormalities on initial presentation (89%) or follow-up MR imaging of the brain (79%).14 The lack of neuroimaging findings in NMDAr encephalitis is consistent across the medical literature, with another study reporting that most patients with NMDAr encephalitis (66%) had normal brain MR imaging findings, and the remaining 44% had wide variation in the distribution and degree of T2-FLAIR hyperintense signal changes throughout the brain.53 Recognizing this established progression of specific symptoms and a lack of neuroimaging findings is essential to prospectively consider the diagnosis in the appropriate clinical setting, particularly when patients demonstrate characteristic electroencephalogram findings.57 When brain MR imaging abnormalities are present, these T2-FLAIR hyperintense lesions can typically demonstrate mild transient cortical enhancement without restricted diffusion or hemorrhage (Fig 4).3,14,53 When brain MR imaging findings are absent but the clinical findings suggest the possibility of an autoimmune encephalitis, brain FDG-PET imaging may be indicated, especially early in the disease process if clinical suspicion for autoimmune encephalitis is high, because it appears to be a more sensitive imaging technique for detecting temporal lobe abnormalities with normal brain MR imaging findings.29,40,44,58
Voltage-Gated Potassium Channel
Voltage-gated potassium channel (VGKC) encephalitis is one of the most common group 2 subtypes of autoimmune encephalitis, which can demonstrate classic features of limbic encephalitis but is primarily defined by the early and prominent development of medically intractable epilepsy.24 The near-universal development of seizures in patients with VGKC encephalitis is partially explained by the high concentration of potassium channels in the limbic structures, and the epileptogenic potential of these antibodies is further supported by the observation that up to 6% of patients with a long-standing history of epilepsy were found to have circulating VGKC autoantibodies.59 It remains unclear whether VGKC antibodies directly contribute to neuronal dysfunction independent of seizure activity, but there is growing consensus that a genetic predisposition to VGKC autoimmunity is probably an independent risk factor for the development of temporal lobe epilepsy.9,59
According to a recent review of 42 patients with VGKC encephalitis, most (69%) demonstrated MR imaging findings classic for autoimmune encephalitis in the acute setting (T2-FLAIR hyperintense lesions in 1 or both medial temporal lobes) and had an increased propensity to develop chronic findings of mesial temporal sclerosis on follow-up imaging (48%).24 A subset of patients with medial temporal lobe lesions demonstrated additional findings of restricted diffusion and postcontrast enhancement (21%) that was highly associated with the development of mesial temporal sclerosis (66%).24 Another important finding was that “extralimbic” involvement in VGKC encephalitis was exceedingly rare (5%).24 The number of specific antibodies within the spectrum of VGKC encephalitis continues to grow, with distinction now being made for antibodies to particular antigens like leucine-rich glioma-inactivated 1, contactin-associated protein-like 2, and dipeptidyl-peptidase-like protein-6, which represent distinct subtypes of autoimmune encephalitis because these antibodies bind not to the Kv1 neuronal antigens of the VGKC but to other juxtaparanodal proteins with a different clinical profile.6,60⇓⇓–63
Voltage-Gated Calcium Channel
Voltage-gated calcium channel (VGCC) encephalitis is a relatively rare subtype described in women and young children, which is associated with the classic clinical progression of symptoms described in group II antibodies (viral prodrome → neuropsychiatric symptoms → limbic dysfunction → seizures) and can have prominent “migratory” extralimbic involvement with gyriform postcontrast enhancement and cortical laminar necrosis (Figs 5 and 6).64,65
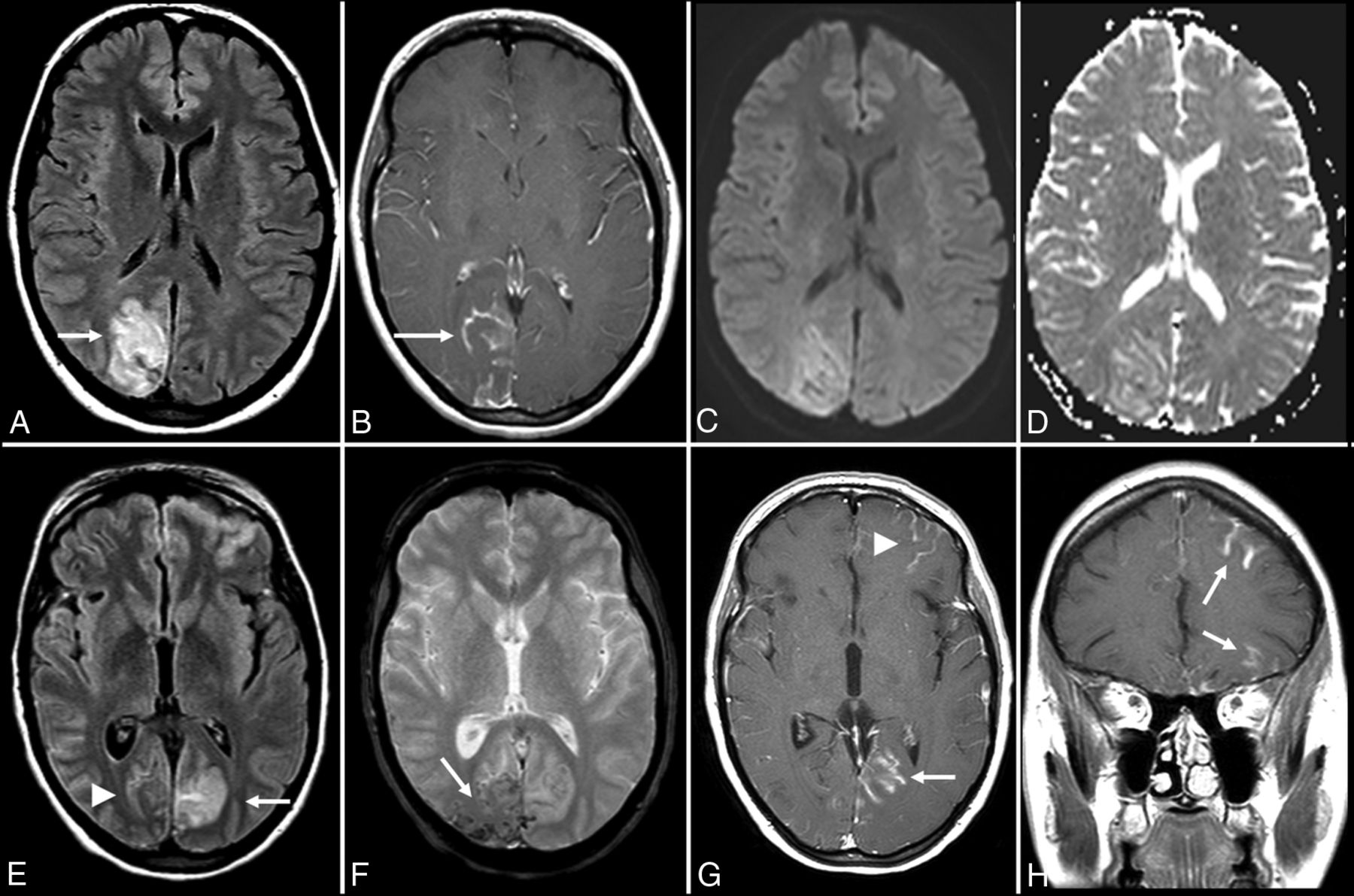
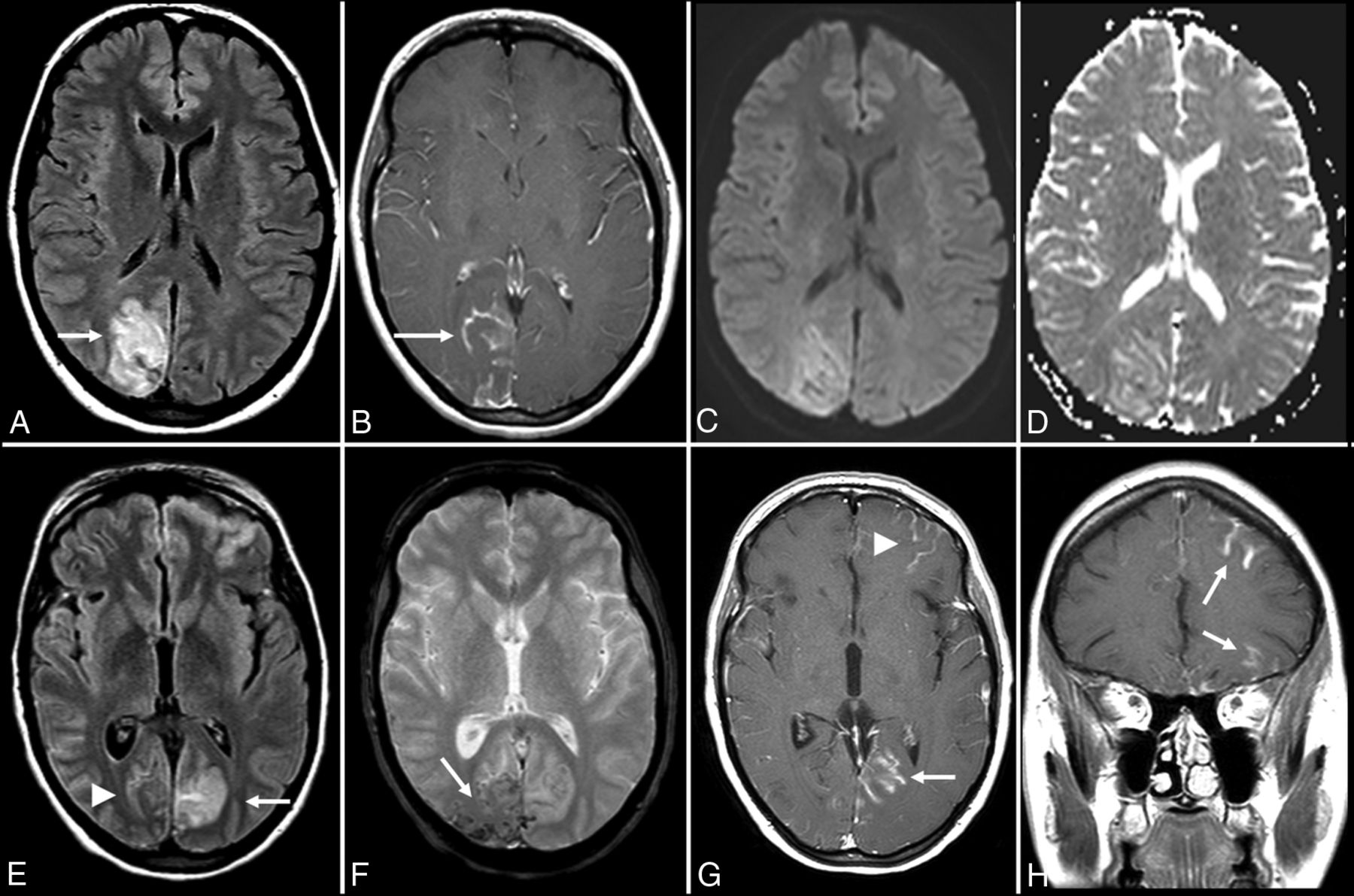
Anti-voltage-gated calcium channel encephalitis. A 39-year-old woman presented with left-sided weakness and left visual field deficits with subsequent development of encephalopathy and seizures. Initial MR imaging of the brain (A–D) demonstrates multifocal T2-FLAIR hyperintense lesions in the right parieto-occipital region (A), with associated pial/sulcal enhancement (B) and mild cortical restricted diffusion and T2 shinethrough within the subcortical white matter on DWI (C) and the corresponding ADC map (D). Follow-up MR imaging of the brain performed 34 days later (E–H) demonstrates decreased T2-FLAIR hyperintensity (E) with cortical laminar necrosis and petechial hemorrhage (F) at the original lesion, with progressive development on subsequent examinations of similar cortical lesions in the contralateral frontal, parietal, and occipital lobes (E–H).

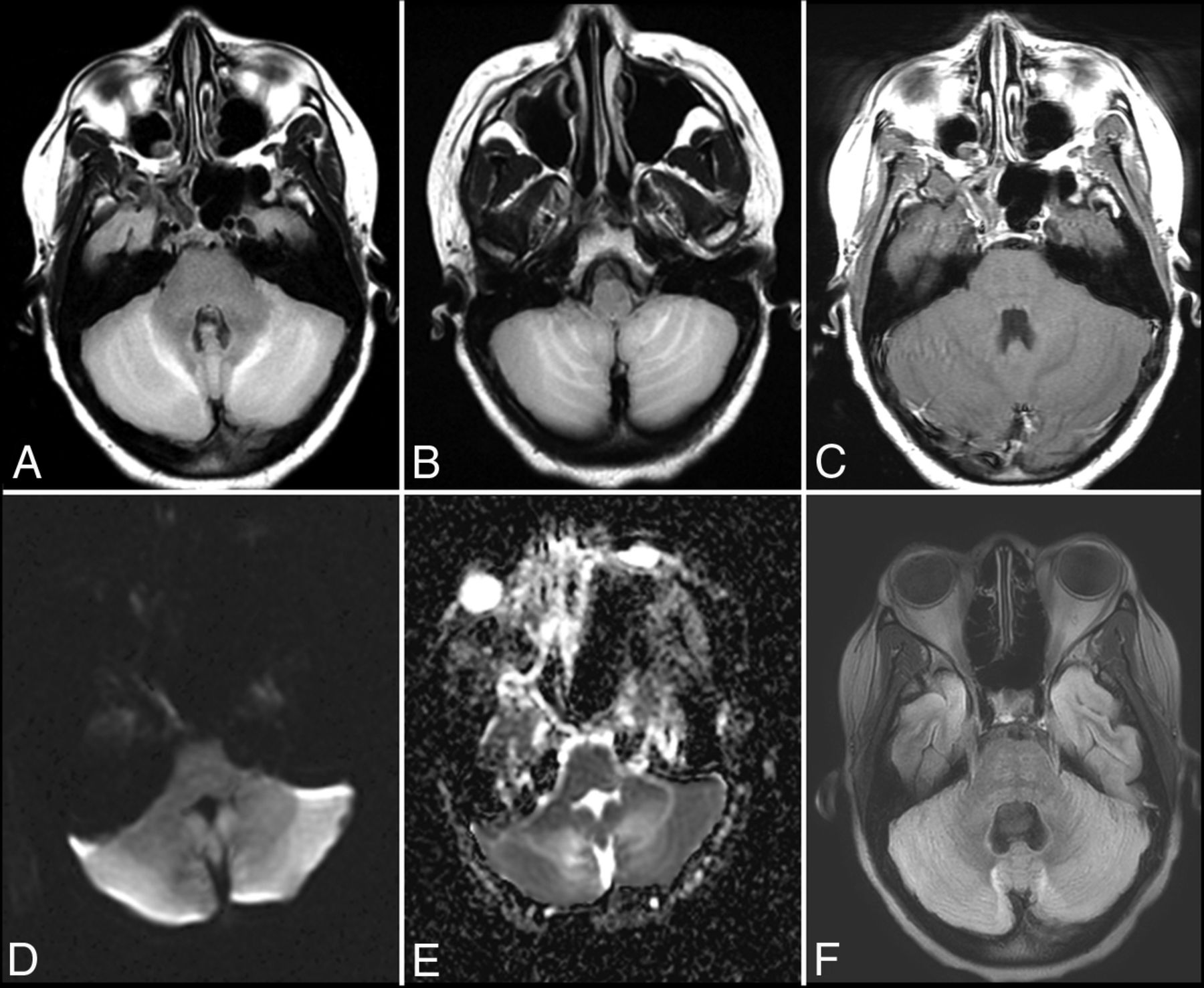
Anti-voltage-gated calcium channel cerebellitis. A 23-year-old woman with a history of autoimmune hepatitis presented with altered mental status. Initial brain MR imaging (A–E) demonstrates T2-FLAIR hyperintensity within the bilateral cerebellar hemispheres with mass effect on the fourth ventricle (A and B), without evidence of postcontrast enhancement (C) and with mild restricted diffusion on DWI (D) and the corresponding ADC map (E). A follow-up scan 1 month later (F) demonstrates resolution of the T2-FLAIR hyperintensity and associated mass effect on the fourth ventricle following a steroid taper.
γ-Aminobutyric Acid Receptor
γ-aminobutyric acid encephalitis (GABAr) has 2 subtypes that both have clinical features similar to those of VGKC encephalitis but are less common and have a better overall prognosis.3,15,43,66 The 2 γ-aminobutyric acid receptor subtypes have different clinical profiles and are characterized by antibodies to either the γ-aminobutyric acid A-receptor or B-receptor subunits.3,15,43,66 Patients with antibodies to the γ-aminobutyric acid B-receptor present with classic features of limbic encephalitis defined by early and frequent seizures with the development of T2-FLAIR hyperintense signal changes in 1 or both temporal lobes.3,43,66 Patients with γ-aminobutyric acid B-receptor antibodies have a higher association with cancer than most other group II antibodies and are more often seen with small-cell lung cancer or pulmonary neuroendocrine tumors.3,43,66 The development of autoimmune encephalitis in these patients usually precedes the diagnosis of cancer but responds well to immunosuppression and removal of the underlying tumor.66 Patients with γ-aminobutyric acid A-receptors also have a good prognosis with adequate treatment, are not associated with cancer, and are unique because in addition to classic MR imaging findings, these patients often demonstrate extensive T2-FLAIR hyperintense lesions outside of the limbic system.3,15
Alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid Receptor
Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAr) encephalitis is an uncommon subtype characterized by the subacute onset of purely psychiatric symptoms with T2-FLAIR hyperintensities isolated to the hippocampus. This subtype has a higher association with cancer than other cell-surface antibody subtypes and is most often seen in women with lung, breast, or thymic tumors.3,67
Additional Type II Antibody Subtypes
Anti-glutamate receptor 3 (GluR3) antibodies have been associated with Rasmussen encephalitis.7 Anti-metabotropic glutamate receptor 1 (mGluR1) antibodies have been described in patients with lymphoma with cerebellar ataxia.7 Anti-metabotropic glutamate receptor 5 (mGluR5) antibodies have been linked to limbic encephalitis associated with Hodgkin lymphoma (Ophelia syndrome).7 Anti-D2 dopamine receptor antibodies represent a rare subtype associated with basal ganglia encephalitis.8 Anti-glyoxylate reductase 1 (GlyR1) antibodies can be seen in 3 related groups distinguished by having dominant clinical features of stiff leg syndrome, stiff person syndrome, or progressive encephalomyelitis with rigidity and myoclonus.6
Systemic Autoimmunity with Encephalopathy
Neuropsychiatric manifestations of systemic autoimmune conditions such as systemic lupus erythematosus can occur and may be mediated by an antibody profile that includes antiphospholipid antibodies and anti-glutamate receptor antibodies.68 Catastrophic antiphospholipid antibody syndrome is a condition that can present with strokelike symptoms and multifocal petechial hemorrhages throughout the brain, which are best seen on susceptibility-weighted MR imaging sequences.68 Patients with thyroid dysfunction and antithyroid antibodies in conditions like Graves disease or Hashimoto thyroiditis can develop encephalopathy associated with their autoimmune thyroid disease that has a characteristic “migratory pattern” of neuroimaging findings with cortical T2-FLAIR lesions in different regions of the brain on sequential MR imaging examinations (Fig 7).11,69 Hashimoto encephalopathy, in particular, is closely associated with autoimmune encephalitis, given its propensity for the combination of encephalopathy, psychiatric symptoms, and seizures.69,70 MR imaging findings in Hashimoto encephalopathy often have more prominent features of leukoencephalopathy (patchy and confluent T2-FLAIR hyperintense lesions in the subcortical, periventricular, and deep white matter).2,43,70
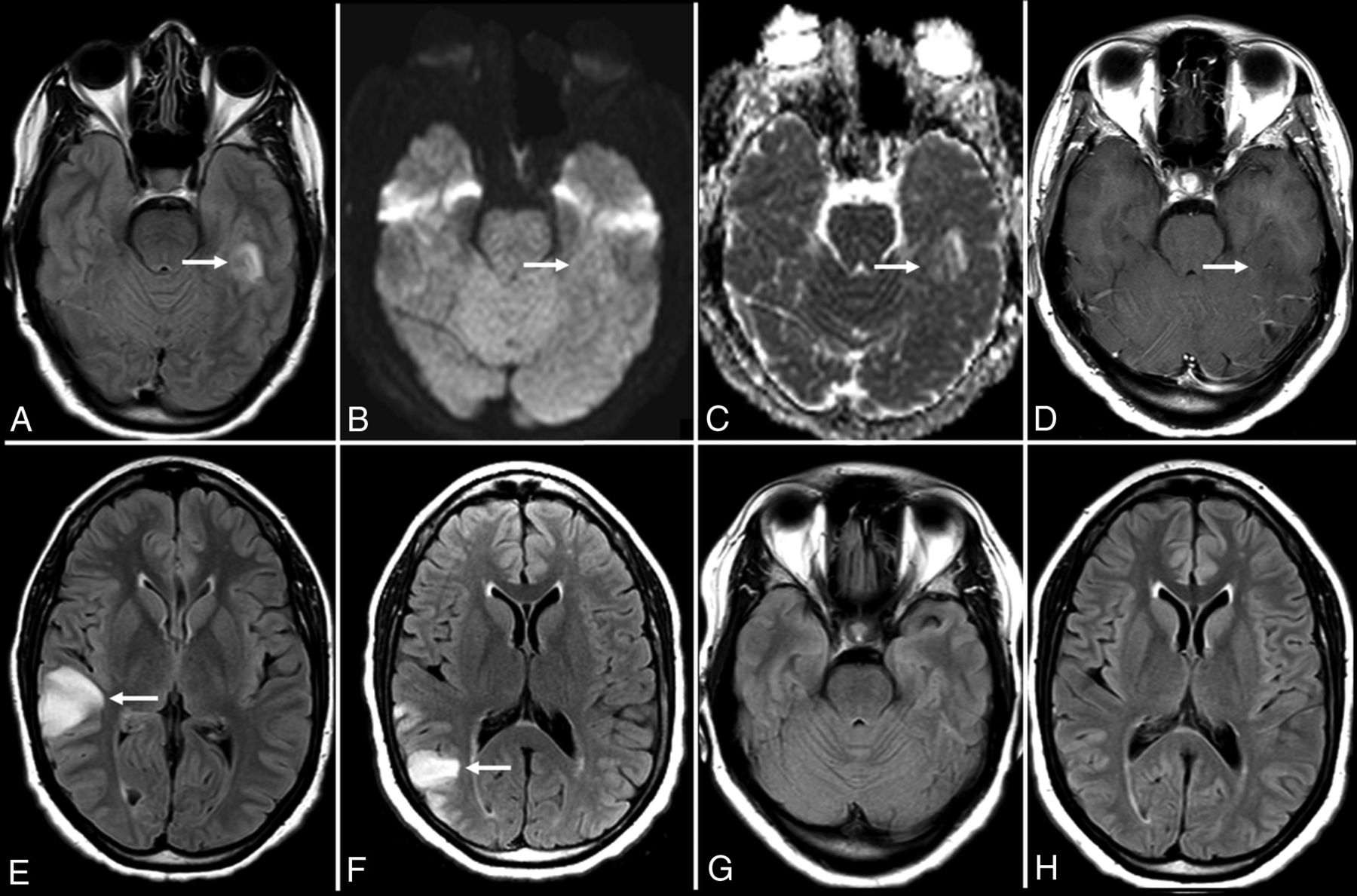
Hashimoto encephalitis. A 41-year-old woman presented with gradually worsening headaches and memory impairment without the development of psychosis or seizures. MR imaging of the brain (time, 0) demonstrates T2-FLAIR hyperintensity in the inferior left temporal lobe (A) without evidence of restricted diffusion (B and C). MR imaging of the brain (time, 21 days) demonstrates enlargement of the lesion on T2-FLAIR (not shown) without postcontrast enhancement (D). MR imaging of the brain (time, 3 months) demonstrates resolution of the prior lesion (not shown) but development of similar T2-FLAIR hyperintensity in the right frontoparietal junction (E). A subsequent scan at approximately 5 months shows near-complete resolution of that lesion with a new T2-FLAIR hyperintense lesion more posteriorly (F). A follow-up scan (G and H) nearly 1 year from onset shows complete resolution of the imaging abnormalities.
Conclusions
Autoimmune encephalitis is an important diagnostic consideration in patients presenting with new onset of altered mental status of unclear etiology. It includes a myriad of clinical conditions that have a common pathophysiology (ie, antibodies directed against CNS structures). The 2 distinct groups (group I, intracellular directed antibodies, and group II, cell-surface directed antibodies) have overlapping clinical and imaging features. Neuroimaging findings will most often involve the limbic structures, but involvement of the striatum, diencephalon, or rhombencephalon can be seen. A subset of patients with autoimmune encephalitis will have no neuroimaging findings despite profound neuropsychiatric dysfunction, but serum antibody testing can still ultimately lead to the diagnosis of autoimmune encephalitis. While there is no single diagnostic feature that can make this diagnosis in isolation, recognizing a certain constellation of findings during the work-up of complex and atypical cases of new-onset altered mental status is crucial to confirm the diagnosis with serologic testing and initiate treatment in a timely fashion.
Acknowledgments
We are grateful to our clinical colleagues in the Department of Neurology within the Henry Ford Health System for their role in diagnosing and treating patients with autoimmune encephalitis at our institution. We would specifically like to acknowledge Dr Daniel Newman, Director of the Henry Ford Hoenselaar ALS Clinic, for his enthusiastic clinical support and expertise. We would also like to thank Dr Chunhai Hao, Chief of Neuropathology in the Department of Pathology and Laboratory Medicine at Henry Ford Hospital, for his work characterizing the histopathology of brain biopsy specimens in cases of suspected autoimmune encephalitis at our institution.
Footnotes
Paper previously presented, in part, at: Annual Meeting of the American Society of Neuroradiology and the Foundation of the ASNR Symposium, May 23–26, 2016; Washington, DC.
Indicates open access to non-subscribers at www.ajnr.org
References
- © 2017 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- MR Imaging Findings in Anti-Leucine-Rich Glioma Inactivated Protein 1 Encephalitis: A Systematic Review and Meta-analysis
- Ovarian teratoma-associated anti-NMDA receptor encephalitis with severe features
- Mimics of Autoimmune Encephalitis: Validation of the 2016 Clinical Autoimmune Encephalitis Criteria
- Teaching NeuroImage: Atypical Unilateral Cortical Ribboning in Anti-NMDA Receptor Encephalitis
- Acute cerebral atrophy in autoimmune encephalitis complicated by haemophagocytic lymphohistiocytosis
- Encephalitis in Previously Healthy Children
- Imaging Review of Paraneoplastic Neurologic Syndromes
- Autoimmune encephalitis in the elderly: who to test and what to test for
- Clinical approach to the diagnosis and successful management of anti-NMDA receptor encephalitis when antibody testing is not possible
- Anti-Amphiphysin-associated limbic encephalitis in a 72-year-old patient with aortic angiosarcoma
- Neuropsychiatric Lupus with Antibody-Mediated Striatal Encephalitis
- Increased prevalence of brain tumors classified as T2 hyperintensities in neurofibromatosis 1