
Abstract
BACKGROUND AND PURPOSE: Autoimmune voltage-gated potassium channel complex encephalitis is a common form of autoimmune encephalitis. Patients with seizures due to this form of encephalitis commonly have medically intractable epilepsy and may require immunotherapy to control seizures. It is important that radiologists recognize imaging characteristics of this type of autoimmune encephalitis and suggest it in the differential diagnosis because this seizure etiology is likely under-recognized. Our purpose was to characterize MR imaging findings in this patient population.
MATERIALS AND METHODS: MR imaging in 42 retrospectively identified patients (22 males; median age, 56 years; age range, 8–79 years) with seizures and voltage-gated potassium channel complex autoantibody seropositivity was evaluated for mesial and extratemporal swelling and/or atrophy, T2 hyperintensity, restricted diffusion, and enhancement. Statistical analysis was performed.
RESULTS: Thirty-three of 42 patients (78.6%) demonstrated enlargement and T2 hyperintensity of mesial temporal lobe structures at some time point. Mesial temporal sclerosis was commonly identified (16/33, 48.5%) at follow-up imaging. Six of 9 patients (66.7%, P = .11) initially demonstrating hippocampal enhancement and 8/13 (61.5%, P = .013) showing hippocampal restricted diffusion progressed to mesial temporal sclerosis. Conversely, in 6 of 33 patients, abnormal imaging findings resolved.
CONCLUSIONS: Autoimmune voltage-gated potassium channel complex encephalitis is frequently manifested as enlargement, T2 hyperintensity, enhancement, and restricted diffusion of the mesial temporal lobe structures in the acute phase. Recognition of these typical imaging findings may help prompt serologic diagnosis, preventing unnecessary invasive procedures and facilitating early institution of immunotherapy. Serial MR imaging may demonstrate resolution or progression of radiologic changes, including development of changes involving the contralateral side and frequent development of mesial temporal sclerosis.
ABBREVIATIONS:
- Ab
- autoantibody
- CASPR2
- contactin-associated protein-like 2
- LGI1
- leucine-rich glioma-inactivated protein 1
- MTS
- mesial temporal sclerosis
- VGKC
- voltage-gated potassium channel complex
Limbic encephalitis, typically characterized clinically by the subacute onset of partial and secondary generalized seizures, memory loss, and neuropsychiatric changes can be of either infectious or autoimmune etiology. Herpes simplex encephalitis is the most common infectious agent that targets the limbic region. Autoimmune limbic encephalitis may occur as a paraneoplastic phenomenon associated with certain tumors or may occur in the absence of a neoplasm.1⇓–3 Autoimmune voltage-gated potassium channel complex (VGKC) encephalitis is one of the most common forms of autoimmune encephalitis. It is frequently found in the absence of associated tumor.4,5 VGKC autoantibodies (Abs) have been reported in 6% of patients with long-standing seizures6 and in up to 11% when both acute and chronic presentations are considered.7 Furthermore, autoimmune VGKC encephalitis may be more common than paraneoplastic autoimmune encephalitis.8 Because the hippocampus and associated limbic structures contain attenuated concentrations of potassium channels and due to the inherent epileptogenic potential of these structures, it is not surprising that alteration of the neuronal electrochemical function by this class of Ab would predispose to seizure activity. Comprehensive information is lacking as to whether VGKC autoimmunity leads to longitudinal loss of neurons in the mesial temporal regions or whether this ion channel Ab disorder is without significant long-term radiologic or histopathologic sequelae. Autoimmune VGKC encephalitis can be diagnosed by serologic testing and is potentially treatable with immunotherapy.5,6,8⇓⇓–11 However, it is suspected that this condition is greatly underdiagnosed and undertreated.9,12 Because the long-term consequences of limbic encephalitis and seizures are frequently significant and the immune aspect of this disease is potentially treatable, it is important that neuroradiologists recognize the existence of this newly identified entity and its imaging characteristics to raise the possibility in the differential diagnosis when appropriate. In this study, we characterize MR imaging findings in autoimmune VGKC encephalitis.
Materials and Methods
Institutional review board approval was obtained, and informed consent was waived for this retrospective review. Patients were identified by the following 2 mechanisms: 1) A search of the Mayo Clinic Life Sciences System (a clinical data repository containing patient demographics, diagnoses, hospital and clinical notes, laboratory results, and pathology data obtained from multiple clinical and hospital source systems) by using the keywords “voltage-gated potassium channel” or “VGKC” and “seizures,” “epilepsy,” “encephalitis” or “encephalopathy” identified 11 patients presenting between January 1, 2001, and June 1, 2008, seropositive for VGKC Ab with associated seizures; 2) From June 1, 2008, to June 30, 2010, comprehensive serologic evaluation of patients with unexplained neurologic symptoms was performed at our institution, identifying 1992 patients who were seropositive for VGKC Ab.13 A search of this data base identified 33 additional patients with associated seizures.
Chart reviews were performed on these 44 patients. All available imaging was reviewed by 2 neuroradiologists with 13 and 12 years of experience, consensus was reached at the time of interpretation, and results were tabulated. Initial MR imaging was evaluated for the presence of T2 signal hyperintensity in the mesial temporal lobes and extratemporal regions, hippocampal and amygdala swelling, restricted diffusion, and abnormal enhancement. All available follow-up imaging, including short-term (≤1 year) and later studies, was assessed for progression or regression of these findings and for the presence of T2 hyperintensity or atrophy indicative of mesial temporal sclerosis (MTS). When MTS was present, the time to its development from the time of first imaging abnormality was recorded.
Statistical Analysis
Statistical analysis was performed by using the JMP software package (Version 9.0; SAS Institute, Cary, North Carolina). χ2 analysis was used to determine the association between the development of MTS and patient age. The Fisher exact test was used to determine the association between the development of MTS and each of the following: sex, enhancement, restricted diffusion, and leucine-rich glioma-inactivated protein 1 (LGI1) or contactin-associated protein-like 2 (CASPR2) subunit status. Only 2-tailed tests were used. A P value of < .05 was considered significant.
Results
Clinical Findings
Clinical results are summarized in the Table. Forty-four VGKC Ab–positive patients with seizures were identified. Two were excluded because their imaging was no longer available for review. MR imaging and clinical findings of the remaining 42 patients (22 males; median age, 56 years; age range, 8–79 years) were reviewed. The clinical aspects of 18 patients in this cohort have previously been published, but that prior report did not include detailed analysis of MR imaging findings.5 Nine patients (21.4%) had malignancies. One (patient 10, with a thymoma) also had concurrent CRMP5 and GAD65 Abs. Thirty-three patients lacked a tumor diagnosis after a median clinical follow-up of 18 months (range, 1 month to 8.5 years). Two patients, both female, presented in the pediatric age range (8 and 16 years of age), and neither had malignancy.
Clinical findings in autoimmune VGKC encephalitis with seizures
All 42 patients had seizures. Thirty-five had seizures at presentation. The other 7 developed seizures 1–6 months after initial neurologic symptom onset. Partial seizures were most common, occurring in 27. Only 1 patient, a 46-year-old woman with alcoholism, developed status epilepticus. In 11 patients, seizure was the only neurologic symptom. Additional neurologic symptoms in the other 31 patients are listed in the Table.
Quantitative VGKC Ab levels were available for 33 patients, and autoantibody subtyping was available in 32. Twenty-two (68.8%) were positive for the LGI1 subtype, 3 (9.4%) were positive for the CASPR2 subtype, and 7 (21.9%) were negative for both subtypes. No patients were positive for both LGI1 and CASPR2 subtypes.
Thirty-two patients (76.2%) were treated with immunotherapy as outlined in the Table. Initiation of treatment ranged from 1 week to 3 years after symptom onset, which varied depending on seizure frequency, clinical response, and clinical course.
Initial MR Imaging Findings
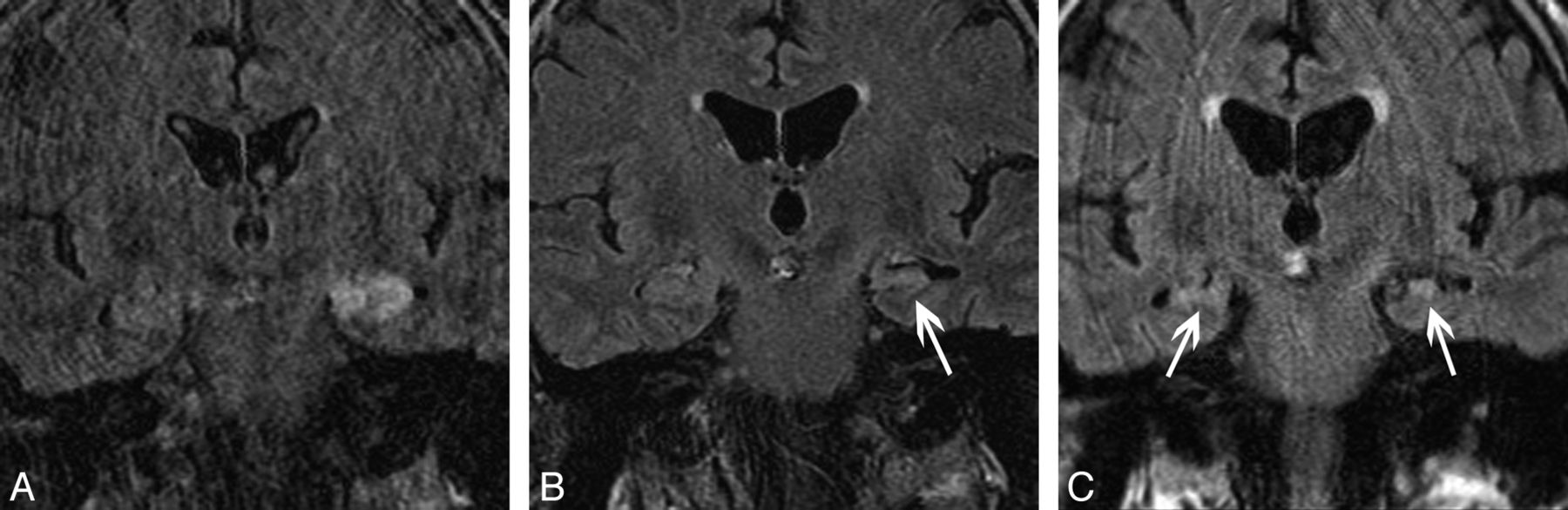
There was variability in the imaging protocol performed during the 10-year span of this study, and in some cases, initial imaging was performed at outside institutions. This resulted in variability in the specific sequences obtained, particularly in the availability of diffusion-weighted and contrast-enhanced images. All patients had T2 and/or FLAIR sequences available for review. Thirteen patients had normal MR imaging findings at presentation. In the 29 (69%) cases with abnormal MR imaging findings at presentation, imaging findings revealed enlargement of the amygdala and/or hippocampus with associated signal hyperintensity on T2-weighted images. Of these, findings were unilateral in 15 patients (35.7%; 9 left-sided; Fig 1A). Findings were bilateral in 14 (33.3%; Fig 2A, -B). In 10 patients post-contrast-enhanced T1-weighted imaging was not performed. In those that had post-contrast-enhanced T1-weighted imaging, 9/32 cases (28.1%) demonstrated mild, ill-defined enhancement in areas of T2 hyperintensity (Fig 2C). In 12 patients, DWI was not performed. In the 30 patients in whom DWI was performed, 13/30 cases (43%) showed associated restricted diffusion in areas of T2 hyperintensity (Fig 3A, -B). Twenty-four patients had both DWI and post-contrast-enhanced T1-weighted imaging; 5 (20.8%) of these patients had both restricted diffusion and abnormal enhancement. No case showed findings of hemorrhage.

A 66-year-old man with autoimmune VGKC epilepsy. Coronal FLAIR at presentation (A), 7-month follow-up (B), and 46-month follow-up (C) demonstrate progression from unilateral increased signal intensity and enlargement of the left hippocampus (A) to left MTS (arrow, B) and finally progression to bilateral MTS (arrows, C).

A 25-year-old man with autoimmune VGKC epilepsy. Imaging at presentation demonstrates enlargement and increased signal intensity in the bilateral hippocampi (A) and bilateral amygdalae (B) on coronal FLAIR, with faint ill-defined enhancement (arrowheads) of the hippocampi (C) on coronal contrast-enhanced T1. Follow-up coronal FLAIR imaging (D) at 4 years demonstrates progression to bilateral MTS (arrows).

A 60-year-old man with autoimmune VGKC epilepsy. Initial imaging findings (not shown) were normal. At 1-month follow-up, axial diffusion-weighted imaging demonstrates restricted diffusion in the left hippocampus (A), left perisylvian cortex (arrowheads), and right caudate nucleus (arrow, B). Axial FLAIR (C) shows enlargement and increased signal intensity of the left hippocampus, and axial contrast-enhanced T1-weighted image (D) shows faint ill-defined enhancement (arrowheads) in the left hippocampus. Coronal FLAIR (E) 3 years after presentation shows progression to left MTS (arrow).
Only 2 patients demonstrated nonmesial temporal findings; in 1, there was T2 hyperintensity and restricted diffusion in the left perisylvian cortex and right caudate nucleus in addition to involvement of the left hippocampus. This patient was previously reported in a case series of VGKC Ab encephalitis mimicking Creutzfeldt-Jakob disease.14 The second case demonstrated T2 hyperintensity and restricted diffusion in the left insula in addition to the left hippocampus. There were no changes in the inferior frontal lobes or cingulate gyrus in any case.
In 1 patient referred to our institution, imaging findings of right hippocampal and amygdala enlargement and T2 hyperintensity had been interpreted as a glioma, leading to temporal lobectomy at the outside institution. Pathology demonstrated moderate-to-marked gliosis with scant chronic perivascular inflammatory T-lymphocyte infiltrates and no evidence of neoplasm.
Follow-Up MR Imaging Findings
Subsequent short-term follow-up studies (<1 year) showed progression to bilateral abnormalities in 28.6% with initially negative (n = 3) or unilateral (n = 5) presentations (Fig 1C). In total, 33 patients (78.6%) demonstrated medial temporal abnormalities at some point during the disease course.
Hippocampal atrophy and T2 hyperintensity consistent with MTS were identified in 16 of 33 patients (48.5%) at follow-up imaging, ranging from 2 to 39 months (median 7 months; 75% quartile 13 months; 25% quartile 3 months) (Fig 4); bilateral in 6 (Fig 1B, -C and Fig 3C) and unilateral in 10. The patients who developed MTS were older (60.25 ± 17.9 years versus 48.9 ± 12.4 years, P = .02) and more likely to be male (n = 12/16, 75%; P = .03). Nine (40.9%, P = .43) patients positive for LGI1 and none positive for CASPR2 developed MTS.

Time to development of MTS from initial MR imaging abnormality.
In patients with initial associated enhancement, 6/9 (66.7%, P = .11) developed MTS, and in patients with initial restricted diffusion, 8/13 (61.5%, P = .03) developed MTS. In all 5 patients who showed both restricted diffusion and abnormal enhancement, there was progression to MTS.
Conversely, in 5 of 33 patients (15.2%), abnormal findings resolved on follow-up imaging with immunotherapy treatment (1–49 months; mean, 19 months; Fig 5). In 1 additional patient, findings resolved without specific treatment. None of the immunotherapy-responsive patients demonstrated enhancement or restricted diffusion at any time point. Nine patients did not have long-term imaging follow-up at our institution, including 4 with initially negative imaging findings.

A 74-year-old man with autoimmune VGKC epilepsy. Coronal FSE T2 image (A) shows mild enlargement and increased signal in the right hippocampus. Follow-up coronal FSE T2 (B) at 1 year shows resolution of hyperintense signal and enlargement of the right hippocampus.
Discussion
Our study demonstrates that initial MR imaging findings associated with autoimmune VGKC encephalitis included uni- or bilateral amygdala and/or hippocampal enlargement and T2 hyperintensity in 78.6% of patients at some time point during the disease course. Nearly half of our patients also demonstrated restricted diffusion; a quarter showed associated mild ill-defined contrast enhancement, and a minority demonstrated extratemporal findings. These findings are nonspecific and are similar to those associated with infectious limbic encephalitis15 and diverse paraneoplastic limbic encephalitides2,16 and to those demonstrated in several smaller series of autoimmune VGKC encephalitis.1,8⇓–10,17,18 Our study emphasizes the need to include autoimmune VGKC encephalitis in the differential diagnosis of patients presenting with these MR imaging findings.
Many VGKC Ab–positive patients can present with seizures and epilepsy as the primary clinical feature in the absence of memory loss and neuropsychiatric symptoms that are frequently present in the full clinical spectrum of limbic encephalitis.5 Furthermore, while some authors have indicated that autoimmune VGKC encephalitis may present as a monophasic illness, others have reported progression, with patients shown to develop bilateral temporal lobe involvement with time.18,19 Our imaging study would support the finding that radiologic progression is common because greater than a quarter (28.6%) of our patients with initially negative study findings or only unilateral findings progressed to bilateral involvement. Whether these changes represent pathology due to persistent CNS inflammation, reflect damage done secondarily by recurrent seizures, or both remains unclear, and prospective longitudinal follow-up will be required to answer this question.
Patients with autoimmune VGKC encephalitis tend to have an unusually high seizure burden5; this may account, in part, for the development of MTS seen in nearly half of our cohort at long-term follow-up. This also implicates VGKC autoimmunity as one potential etiology for “cryptogenic” MTS. Radiologic features of MTS are without a specific pathologic cause, and investigation for VGKC autoimmunity has not been reported in idiopathic MTS.20 Our findings would argue for the need to consider VGKC autoimmunity as a pathologic mechanism of MTS among such patients. In our cohort, patients who developed MTS were significantly more likely to be male and older than those who were responsive to immunotherapy. Most who developed MTS did so within a year of initial imaging abnormality.
While diffusion-weighted imaging was not included on all initial MR imaging examinations, a statistically significant number of patients with initial imaging findings of T2 hyperintensity and swelling of the mesial temporal lobe structures and associated restricted diffusion progressed to MTS. Restricted diffusion can be seen with recent seizure activity or status epilepticus.21,22 Only 1 of our patients with restricted diffusion had status epilepticus. Unfortunately, this patient did not have long-term follow-up imaging at our institution. Restricted diffusion is more commonly seen in ischemia/infarction and is reflective of cytotoxic edema, typically indicative of irreversible cell damage. Perhaps patients with autoimmune VGKC encephalitis who present with restricted diffusion should be targeted for more prompt or aggressive immunotherapy in an effort to prevent progression to MTS.
Similarly, there was a trend to develop MTS in two-thirds of patients whose scans demonstrated mild ill-defined enhancement in the hippocampus. This suggests that enhancement and its presumed association with breakdown of the blood-brain barrier may be a marker for more severe or irreversible damage to the hippocampus. This was not statistically significant, likely secondary to the small number of patients with enhancement and follow-up imaging available for review at our institution. Potential association of contrast enhancement and ultimate development of MTS should be confirmed with prospective studies involving larger numbers of patients. All 5 patients who had both contrast-enhancement and restricted diffusion went on to develop MTS. Therefore, we suggest that patients imaged for suspected autoimmune limbic encephalitis should have diffusion-weighed imaging and receive contrast as part of the imaging work-up when issues such as impaired renal function or pregnancy do not preclude its use.
T2 hyperintensity and enlargement were confined to the hippocampus and/or amygdala in all except 2 cases in this series, findings that would be atypical for other etiologies of limbic encephalitis. Nevertheless, the imaging findings of our VGKC Ab–positive patients were not entirely specific for limbic encephalitis in all cases. Consideration for glioma, herpes simplex encephalitis, infarction, and Creutzfeldt-Jakob disease delayed diagnosis in some. One of our patients had temporal lobectomy performed at an outside institution for suspicion of glioma, further supporting the value of assessing for VGKC Ab. Only 1 patient demonstrated extratemporal findings of restricted diffusion in a distribution similar to that of Creutzfeldt-Jakob disease.14 There was no evidence of hemorrhage in any case in our series as may be seen in herpes encephalitis.
Interpretation of longitudinal imaging findings in this study was complicated by several factors. First is the long time span during which imaging was obtained, from January 2001 to June 2010. During this decade-long period, the imaging equipment and quality of imaging changed, potentially casting doubts on the consistency of the imaging evaluations. This difference may have underestimated the detection of findings on poorer quality examinations performed at earlier time points. Furthermore, imaging findings are presumed to be reflecting a dynamic pathologic process, likely reflecting not only the primary pathologic autoimmune inflammatory process but also findings associated with engendered seizure activity and those of a superimposed response to immunotherapy and antiepileptic drug treatments. Due to the retrospective nature of our study, we did not specifically assess differences in seizure burden in patients who developed MTS versus those who did not. We propose that future prospective studies may be helpful in addressing this question. Finally, due to the retrospective nature of our study, in some cases, it was difficult to temporally relate treatment regimens with specific imaging findings. However, imaging findings resolved in 14.7% of our patients with some form of immunotherapy treatment. Future prospective studies are anticipated to permit more discrete analysis of the various contributors to the evolution of imaging findings.
Conclusions
The most common initial findings in autoimmune VGKC encephalitis with seizures are T2 hyperintensity and swelling of the limbic structures, though extratemporal involvement does not preclude a diagnosis of this condition. Recognition of these imaging findings and inclusion of autoimmune VGKC encephalitis in the differential diagnosis with other forms of limbic encephalitis may prompt appropriate serologic evaluation to confirm the diagnosis, which may in turn facilitate early institution of immunotherapy and prevent unnecessary invasive procedures. This study also demonstrates that serial MRI should be considered in patients with suspected autoimmune VGKC epilepsy, to demonstrate the following: delayed development of radiologic changes in patients who previously had normal MR imaging findings; evolution of changes involving the contralateral side, suggesting ongoing inflammatory activity radiologically; and progression to MTS. Future prospective work should be performed to answer the important question of how commonly otherwise “cryptogenic” MTS relates to a remote effect of VGKC autoimmunity.
Footnotes
Disclosures: Sean J. Pittock—UNRELATED: Consultancy: Alexian Pharmaceuticals,* MedImmune,* Grants/Grants Pending: RO1 NS065829,* Guthy-Jackson Charitable Foundation,* Payment for Lectures (including service on Speakers Bureaus): American Academy of Neurology,* American Neurological Association,* Patents (planned, pending or issued): patents relating to AQP4 antibodies for the diagnosis of neuromyelitis optica and AQP4 autoantibody as a cancer marker. *Money paid to the institution.
Paper previously presented in part at: 49th Annual Meeting of the American Society of Neuroradiology, June 4–9, 2011; Seattle, Washington.
REFERENCES
- Received February 5, 2013.
- Accepted after revision March 19, 2013.
- © 2014 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Mesiotemporal Volumetry, Cortical Thickness, and Neuropsychological Deficits in the Long-term Course of Limbic Encephalitis
- Clinical Course and Features of Seizures Associated With LGI1-Antibody Encephalitis
- Network-wide abnormalities explain memory variability in hippocampal amnesia
- Rapidly progressive global cerebral atrophy in the setting of anti-LGI1 encephalitis
- Beyond the limbic system: disruption and functional compensation of large-scale brain networks in patients with anti-LGI1 encephalitis
- LGI1, CASPR2 and related antibodies: a molecular evolution of the phenotypes
- Autoimmune Encephalitis: Pathophysiology and Imaging Review of an Overlooked Diagnosis
- Anti-LGI1 encephalitis: Clinical syndrome and long-term follow-up
- Neuronal autoantibodies in mesial temporal lobe epilepsy with hippocampal sclerosis
- Recognizing Autoimmune-Mediated Encephalitis in the Differential Diagnosis of Limbic Disorders
- Basal ganglia T1 hyperintensity in LGI1-autoantibody faciobrachial dystonic seizures