
Abstract
SUMMARY: Ataxia is the principal symptom of many common neurologic diseases in childhood. Ataxias caused by dysfunction of the cerebellum occur in acute, intermittent, and progressive disorders. Most of the chronic progressive processes are secondary to degenerative and metabolic diseases. In addition, congenital malformation of the midbrain and hindbrain can also be present, with posterior fossa symptoms related to ataxia. Brain MR imaging is the most accurate imaging technique to investigate these patients, and imaging abnormalities include size, shape, and/or signal of the brain stem and/or cerebellum. Supratentorial and cord lesions are also common. This review will discuss a pattern-recognition approach to inherited cerebellar ataxia in childhood. The purpose is to provide a comprehensive discussion that ultimately could help neuroradiologists better manage this important topic in pediatric neurology.
ABBREVIATIONS:
- AR
- autosomal recessive
- CAC
- cerebellar ataxia in childhood
- 4H
- hypomyelination with hypogonadotropic hypogonadism and hypodontia
- JSRD
- Joubert syndrome and related disorders
- OPHN1
- oligophrenin-1
Ataxia is an inability to coordinate voluntary muscle movements that cannot be attributed to weakness or involuntary muscle activity.1 It may be present in multiple parts of the body, leading to disorders of gait, limb, or trunk control. Although gait ataxia can be caused by peripheral sensory impairment, it is often observed in association with dysfunction or lesions of the cerebellum or its afferent or efferent tracts. In this context, it is typically accompanied by abnormal eye movements, nystagmus, dysarthria, dysmetria, or dysdiadochokinesia.
CAC can be caused by acquired or inherited diseases, including brain injury (stroke, trauma, hypoxia-ischemia), malformation (Joubert syndrome and associated disorders, cerebellar dysgenesis, rhombencephalosyanapsis), hereditary degenerative disorders (ataxia-telangiectasia, Friedreich ataxia, spinocerebellar ataxias, Marinesco-Sjögren syndrome), metabolic disorders (lipidoses, mitochondrial disorders, lysosomal storage disorders, neuronal ceroid lipofuscinosis, maple syrup urine disease), neoplastic/paraneoplastic disorders, immune-mediated/demyelinating disorders, and drugs/toxins (antiepileptic medications, antihistamines, barbiturates, heavy metals [lead, mercury]).2 Inherited CAC is composed of an extremely heterogeneous group of disorders classified in the literature by many terms, including AR cerebellar ataxias, early-onset cerebellar ataxias, or AR spinocerebellar ataxias.3⇓⇓⇓⇓⇓⇓⇓–11 The mode of inheritance can be AR (most common, usually of early-onset), autosomal dominant (typically adult-onset), X-linked, or maternally inherited forms.
The absolute prevalence of inherited CAC is not known, though it has been estimated to be 5.3 per 100,000 inhabitants in some regions.12 Although few causes of inherited CAC can be treated, a specific diagnosis is important for accurate genetic counseling because many of the disorders are inherited as AR traits and the risk of having an affected child in future pregnancies is 25%. In addition, new drugs and gene and stem cell therapies are being developed and will require a better understanding and a confident diagnosis of the underlying disease.
For clinicians, inherited CACs present a diagnostic challenge because patients typically present during a time of rapid neurologic development; differentiation of a static deficit from a progressive one is challenging when the baseline is undergoing normal change.3⇓–5 In addition, the many recently created diagnostic tools make it difficult for clinicians to determine what investigations to undertake and in which order. A stepwise clinical approach to CAC is necessary to reduce the list of diagnostic possibilities; factors to consider, including mode of presentation, age of onset, familial history, and clinical findings, can help clinicians to tailor the work-up to specific biochemical tests, enzyme activities, mutation analysis, electrophysiologic examination, and imaging.
MR imaging is an excellent tool for assessing the anatomic and biochemical disturbances of the brain in patients with CAC.13⇓⇓⇓–17 It can define structural disturbances; distinguish acute from subacute, chronic, or inherited diseases; suggest some specific diagnoses while eliminating others; and monitor disease progression. Currently, the strength of neuroimaging in diagnostic investigations of many childhood neurologic disorders is based on a pattern-recognition approach, similar to those used in assessing leukodystrophies and brain tumors. Although attention has begun to focus on the embryology, genetics, and imaging of metabolic disorders of the brain and congenital midbrain and hindbrain malformations,15,16,18,19 a pattern-recognition approach to childhood ataxia is lacking. As a result, the neuroradiologist may have difficulty suggesting a concise list of possible diagnoses to the pediatric neurologist or geneticist, even when the MR imaging findings are clearly abnormal. The problem is further compounded by the use of somewhat imprecise terms, such as “pontocerebellar hypoplasia,” which is sometimes used descriptively and other times as a specific diagnosis,20 adding further confusion to the work-up of these patients.
In this imaging-oriented approach to CAC, the more common diseases that cause childhood ataxia have been separated into 2 main categories, degenerative and malformative patterns, on the basis of the imaging appearance. The degenerative pattern is characterized by volume loss (cerebellar atrophy), often associated with WM or gray matter T2/FLAIR signal changes. Most of these disorders are generally considered to be degenerative ones or inborn errors of metabolism (the distinction between these 2 categories is rather vague), and some are treatable. The malformative pattern is characterized by abnormal morphology of the brain stem and/or cerebellum (usually the cerebellar cortex); the structures may be dysmorphic or hypoplastic, with folia and fissures being abnormally shaped rather than merely having an abnormal size.
Because we are focused on inherited diseases, we will not discuss the acute and acquired causes of cerebellar ataxia (such as those due to injury, toxic exposure, radiation, or brain tumor); imaging is usually quite specific in tumors, while clinical history is most useful to establish a diagnosis in the other causes. Because the absolute prevalence of most disorders with CAC is not known, we list the most prevalent diseases based on literature data and the authors' experience.12⇓–14 It is beyond the scope of this article to review all potential causes of childhood ataxia. Our goal is to emphasize MR imaging patterns, clinical findings, and confirmatory tests that the well-informed neuroradiologist can suggest, thus playing an important role in the early management of affected patients.
Degenerative Pattern of CAC
Children with CAC characterized by the degenerative pattern usually present with progressive or intermittent ataxia after the early infantile period (On-line Table 1). Some diseases from this group can be treated; therefore, a vigorous work-up should be performed to find a diagnosis and begin therapy as soon as possible. According to the literature, the most common causes of CAC with cerebellar atrophy are Friedrich ataxia, ataxia with oculomotor apraxia types 1 and 2, ataxia telangiectasia, and infantile-onset spinocerebellar ataxia.3,4,9,12,14 Clinical clues for a conclusive diagnosis can be found from history, physical examination (in ataxia telangiectasia), and laboratory examinations, but imaging is usually requested as a complementary test.
Typical MR imaging abnormalities in these patients are cerebellar atrophy and signal changes in the cerebellum, brain stem, or both. Reduced diffusivity and abnormal MR spectroscopy peaks can also be present. However, the most common abnormality in this group is cerebellar atrophy (Fig 1), a diagnosis that can be made when the cerebellar cortex is thin, resulting in shrunken folia and large cerebellar fissures, or if the cerebellar volume has been shown to progressively diminish on sequential scans.13,14,21,22 Cerebellar atrophy can be global or may be restricted to the vermis or hemispheres; most commonly, it is more pronounced in the vermis. The atrophy can vary from mild to severe, depending on the specific cause, disease stage, genotype, and other factors (Fig 2). Although some reports suggest that certain diseases preferentially affect ≥1 specific lobe of the vermis (eg, the superior vermis is predominately affected in AR ataxia of Charlevoix-Saguenay), our experience suggests that the entire vermis is compromised in most cases. Unilateral cerebellar atrophy in childhood is usually not genetic, and acquired causes should be ruled out first, including fetal injury (such as ischemic stroke or hemorrhage); close examination will show irregular thinning or disrupted foliation of the cortex or a frank cleft. Less common causes, clinical findings, and additional MR imaging features are listed in On-line Table 1.
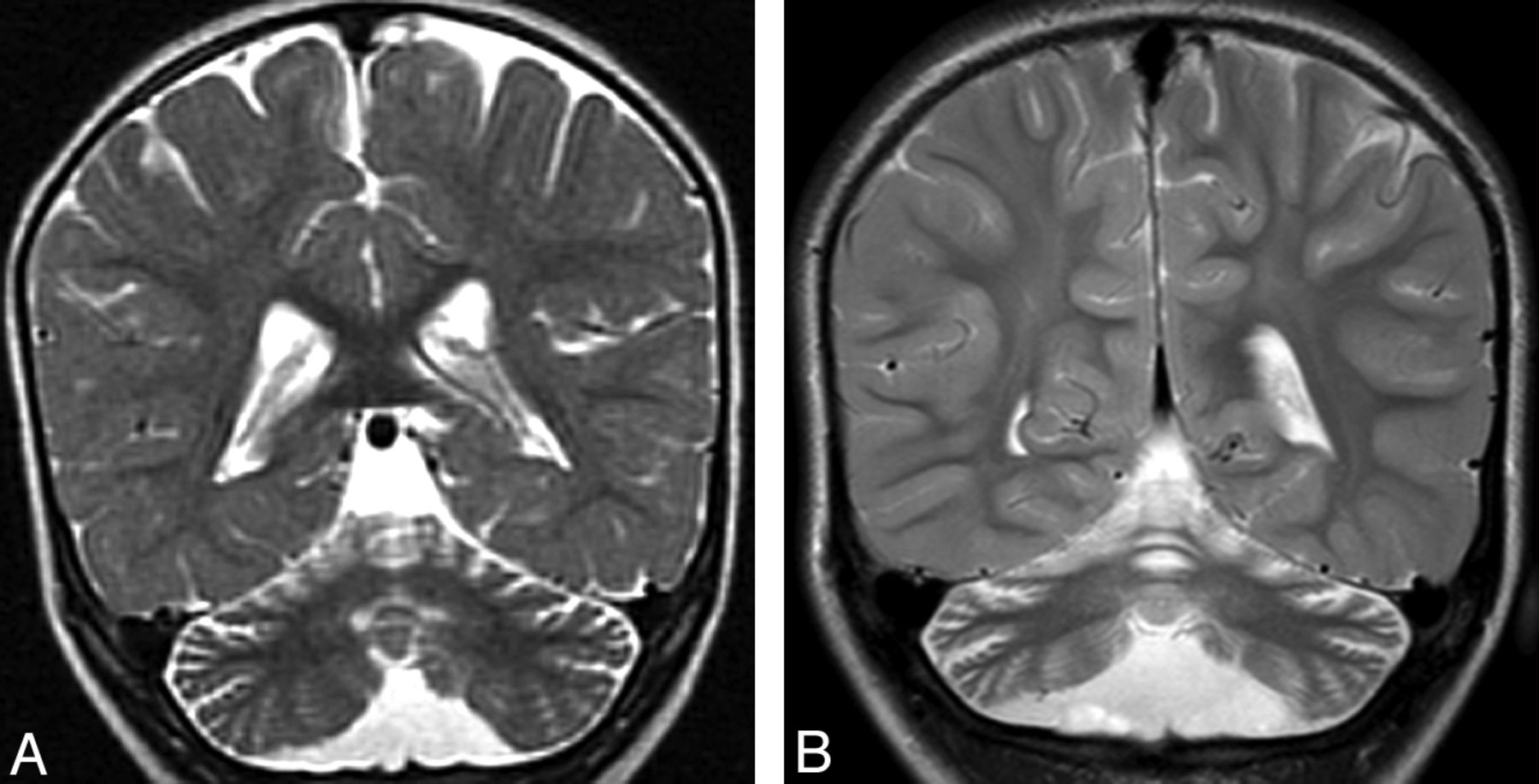
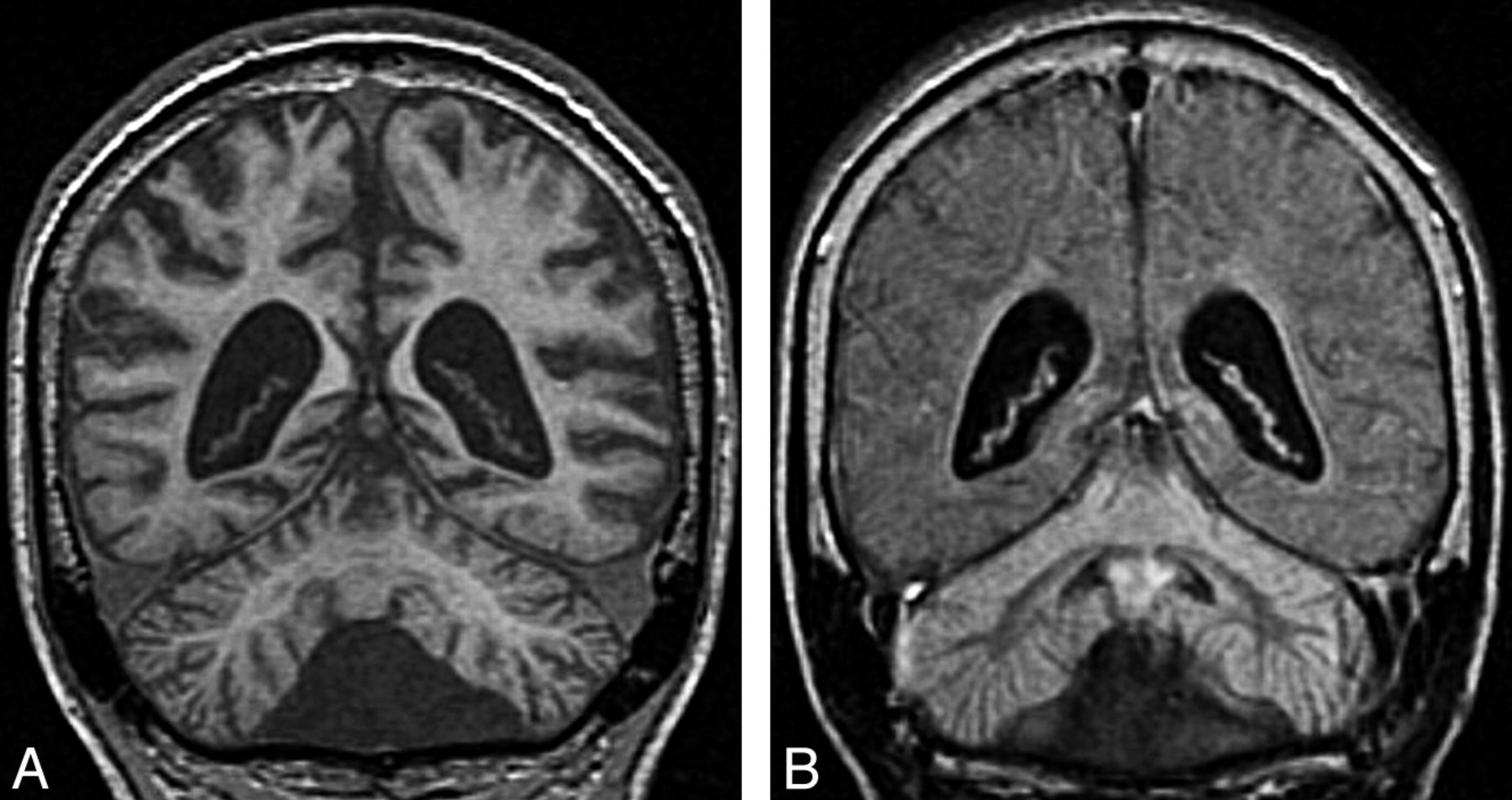
Cerebellar atrophy. A, Coronal T2 image shows cortical thinning and loss of underlying white matter leading to enlarged fissures in the vermis and cerebellar hemispheres. B, Follow-up coronal T2 image 3 years later shows progressive cortical and white matter atrophy.
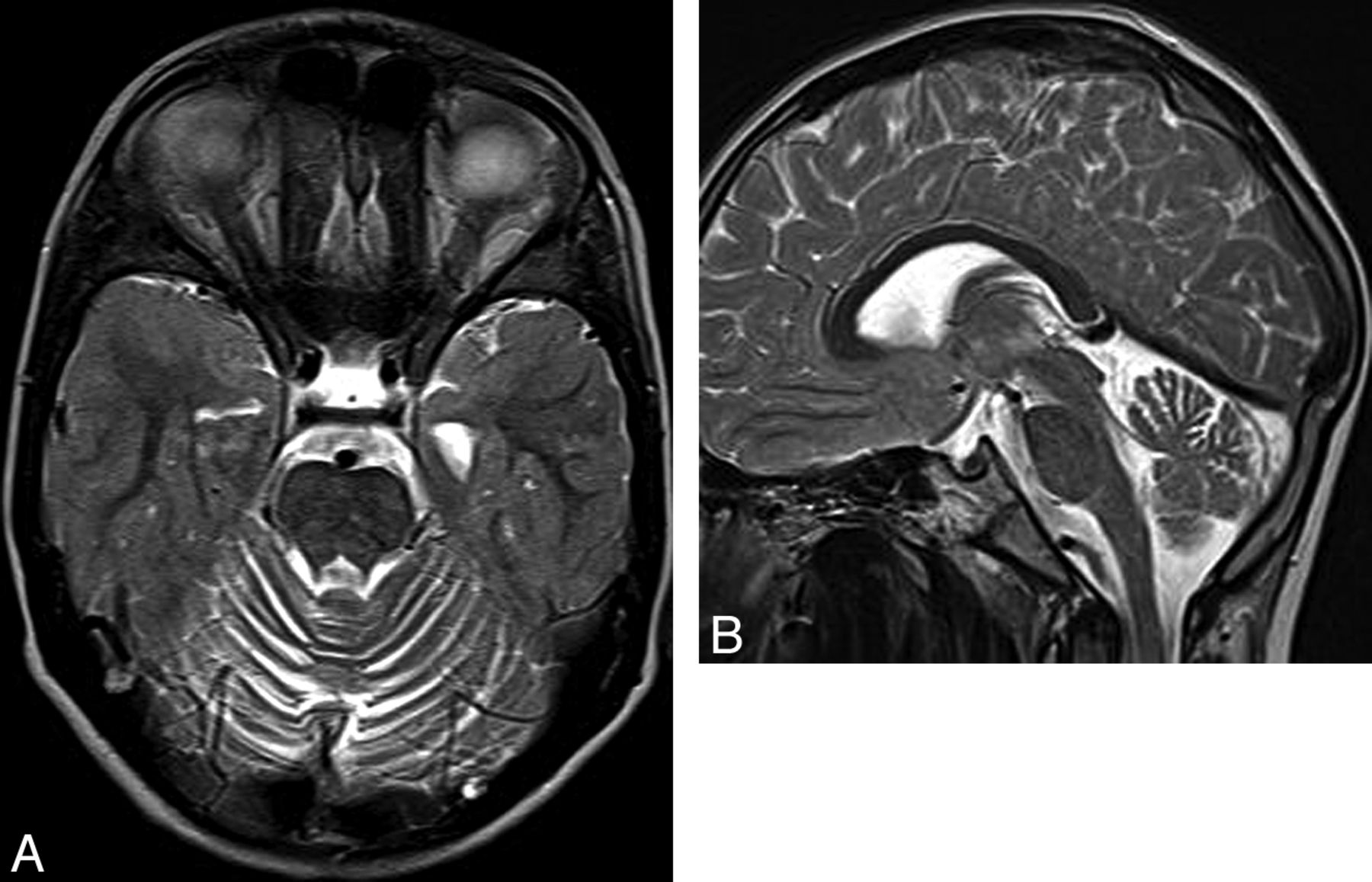
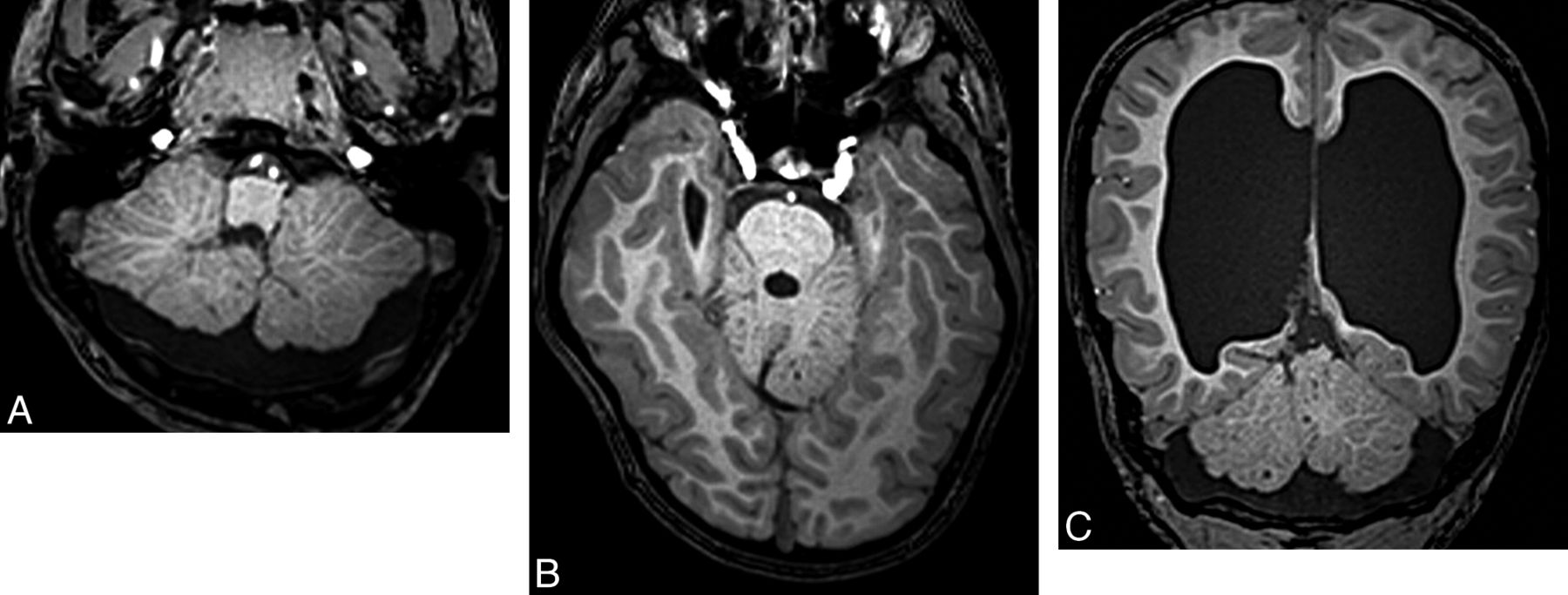
Ataxia-telangiectasia. A, Axial T2 image shows cortical thinning and fissure enlargement in the vermis and cerebellar hemispheres. B, Midline sagittal T2 image demonstrates an atrophic vermis but normal volume of the brain stem. Although nonspecific, this pattern of diffuse cerebellar atrophy suggests a degenerative disease.
Most degenerative causes of CAC are not associated with noticeable brain stem atrophy (Fig 2). This feature probably reflects the concept that an early neuronal injury in the developing cerebellum causes pontine hypoplasia by reducing the number of crossing pontine axons, a phenomenon that usually does not occur in the later degenerative causes of CAC (because the fibers have already crossed and formed synapses with pontine nuclei).16,18 Some degenerative diseases, such as infantile-onset spinocerebellar ataxia and dentatorubral-pallidoluysian atrophy, seem to be exceptions to this rule, however, possibly because of direct involvement of the brain stem in addition to the cerebellum.13
Changes in signal intensity seem to be less common than atrophy in CAC. When present, the signal abnormalities usually involve predominantly the WM and appear as T2/FLAIR hyperintensity on MR imaging, especially in patients with dentatorubral-pallidoluysian atrophy, cerebrotendinous xanthomatosis, infantile Refsum disease, and adrenomyeloneuropathy.13,23⇓–25 Cerebellar gray matter signal changes are uncommon but, when present, should suggest infantile neuroaxonal dystrophy, late infantile neuronal ceroid lipofuscinosis, mitochondrial disorders, or Marinesco-Sjögren syndrome (Fig 3).
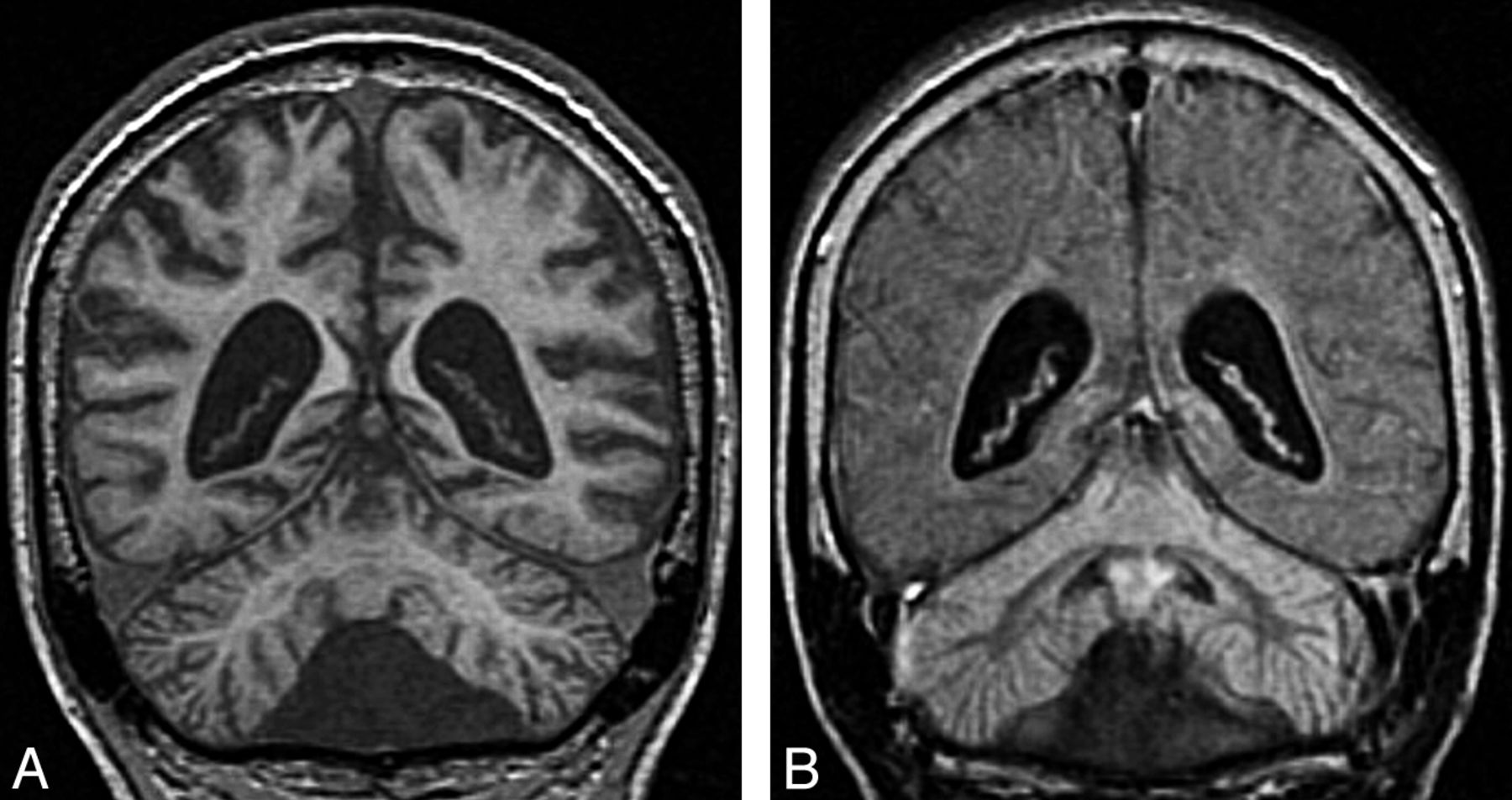
Infantile neuronal ceroid lipofuscinosis. Coronal T1 image (A) shows cerebral and cerebellar atrophy. Coronal FLAIR image (B) demonstrates diffuse abnormal signal intensity affecting the cortex of both cerebellar hemispheres. Compare the signal intensity of the cerebellar cortex with occipital cortex.
In an acute crisis (such as metabolic decompensation from mitochondriopathies, aminoacidurias, or organic acidurias), affected gray matter or WM may show mild edema (swelling with T1 hypointensity and T2/FLAIR hyperintensity) and reduced diffusivity on MR imaging (Fig 4).13 Chronically, the involved structures show volume loss with or without T2 hyperintensity, depending on many factors such as the cause, disease phase, timing of the onset of treatment, and imaging sequences/parameters.

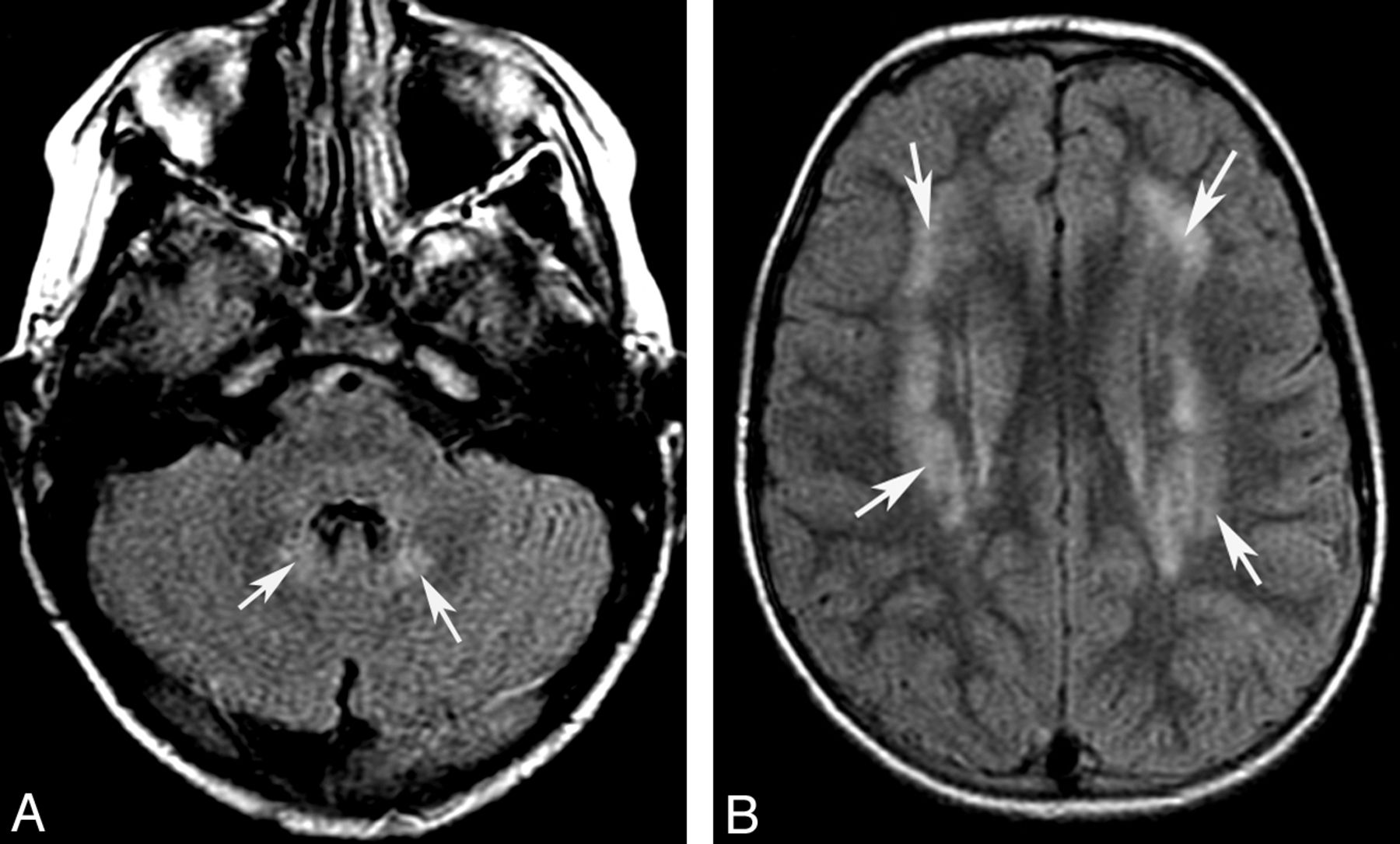
Acute stage of maple syrup disease. Axial T2 images (A and B) show diffuse signal change affecting deep cerebellar white matter (arrows) and midbrain (arrowheads). Trace from diffusion-weighted imaging shows reduced diffusivity in several regions of the midbrain (C).
Although unusual, symmetric T2 hypointensity in the deep cerebellar nuclei (from medial to lateral, the fastigial, globiform, emboliform, and dentate nuclei) has been described in some patients with cerebrotendinous xanthomatosis (Fig 5), reflecting the deposition of hemosiderin, small hemorrhages, and focal calcifications described in histopathologic studies.25 Recently, some reports have suggested that iron deposition and consequent T2 hypointensity resulting from magnetic susceptibility differences can occur in Friedrich ataxia, and a pattern of linear T2 hypointensity in the pons was described in patients with AR ataxia of Charlevoix-Saguenay.16,26⇓⇓–29 If T1 hyperintensity of the globus pallidus is associated with childhood ataxia, Wilson disease should be considered.

Cerebrotendinous xanthomatosis. Axial T2 image shows signal change affecting the deep cerebellar white matter (arrows). Notice the abnormal hypointensity in the gray matter of the cerebellar nuclei (arrowheads).
The list of leukodystrophies that can present with ataxia as a symptom is practically endless (see note in On-line Table 1), and additional supratentorial findings should be systematically evaluated, searching particularly for hypomyelination or abnormal signal intensity in the WM or basal ganglia. The finding of hypomyelination in a child with ataxia should raise suspicion for hypomyelination with atrophy of the basal ganglia and cerebellum (look for small basal ganglia and cerebellar atrophy), 4H syndrome (cerebellar atrophy with normal basal ganglia), and hypomyelination with congenital cataracts (look for cataracts or lens replacements in a child) (Fig 6).30,31 More rarely, ataxia is a predominant symptom of other hypomyelinating disorders such as Pelizaeus-Merzbacher disease, Salla disease, or fucosidosis.31

Hypomyelination with congenital cataracts. Axial FLAIR images (A and B) demonstrate diffuse signal-intensity abnormality (arrows) in the cerebellar (A) and cerebral (B) white matter.
Another feature that may be useful is identification of the involvement of specific white matter tracts. If the corticospinal tracts are specifically affected, peroxisomal diseases (X-linked adrenoleukodystrophy, adrenomyeloneuropathy, and acyl-coenzyme A oxidase deficiency), cerebrotendinous xanthomatosis, and Refsum disease should be considered (Fig 7). If lesions occur in the dorsal spinal cord, the differential diagnosis should include Friedrich ataxia, ataxia with vitamin E deficiency, abetalipoproteinemia, and posterior column ataxia with retinitis pigmentosa.13,32 Abnormal T2/FLAIR hyperintensities in the brain stem suggest Wilson disease (predominantly midbrain), mitochondrial disorders (cerebral peduncles, periaqueductal gray matter, or dorsal pons), juvenile onset of Alexander disease (mostly dorsal medulla), dentatorubral-pallidoluysian atrophy, or cerebrotendinous xanthomatosis (Fig 8).

Adrenoleukodystrophy (A and B) and peroxisomal acyl-coenzyme A oxidase deficiency (C). Axial T2 (A) shows pontine corticospinal tract lesions (arrows), and postcontrast axial T1 (B) demonstrates bilateral and symmetric white matter demyelination and inflammation (enhancement, arrowheads) involving the parietal lobes and splenium of the corpus callosum. Cerebellar white matter and pontine corticospinal tract lesions (C) are also shown in a patient with peroxisomal acyl-coenzyme A oxidase deficiency.
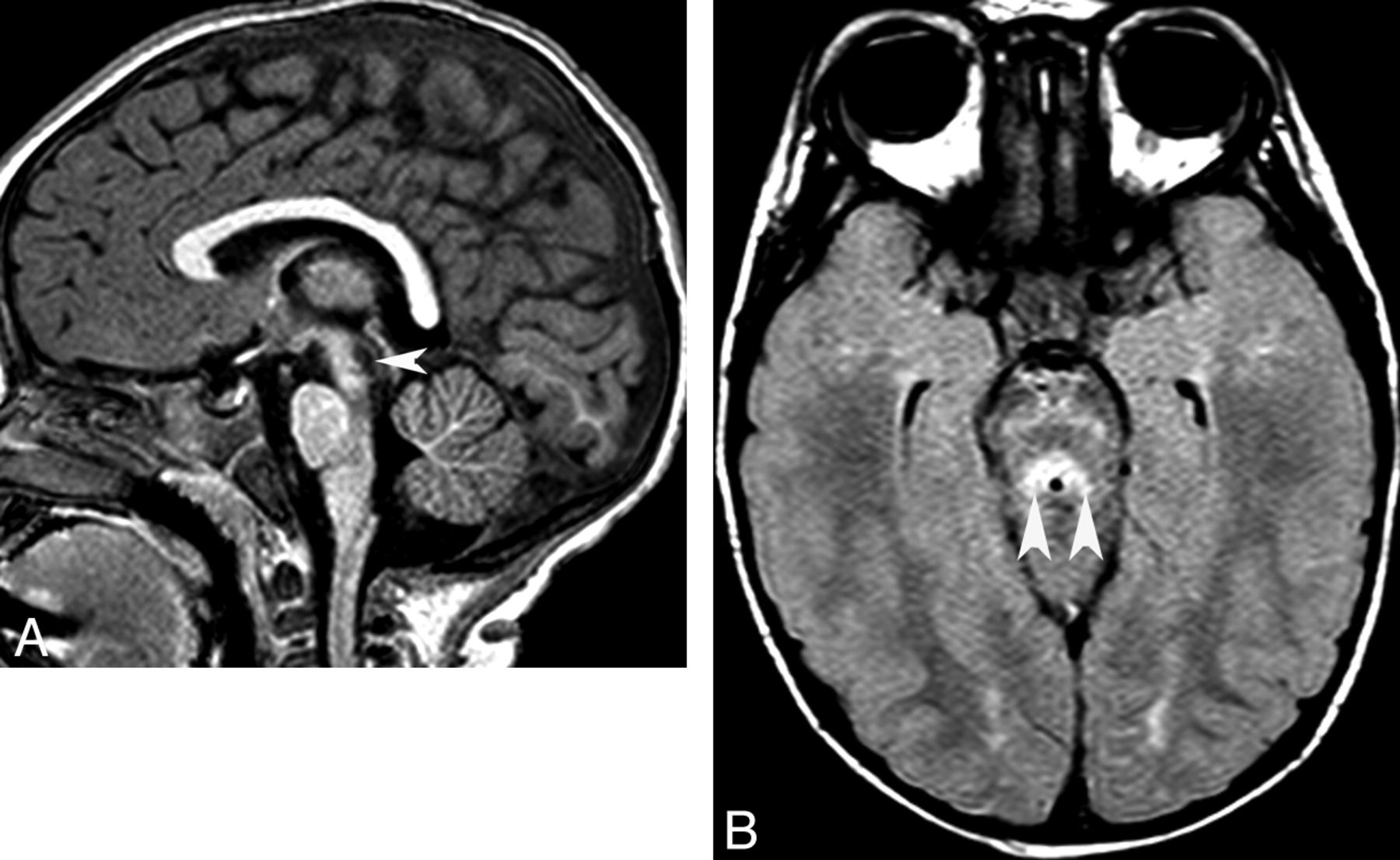
Mitochondrial disorder. Sagittal T1 (A) and axial FLAIR (B) images show abnormal signal intensity (arrowheads) in the dorsal midbrain.
Another characteristic pattern is T2/FLAIR hyperintensity in the hila of the dentate nuclei, which, in the setting of CAC, should suggest cerebrotendinous xanthomatosis, Refsum disease, mitochondrial diseases (Figs 5 and 9), or L-2-hydroxyglutaric aciduria. L-2-hydroxyglutaric aciduria can present with a slowly progressive ataxia. It is an AR disorder caused by mutations of the L2HGDH gene; MR imaging shows T2/FLAIR hyperintensity in the supratentorial subcortical WM, basal ganglia, and cerebellar nuclei.33
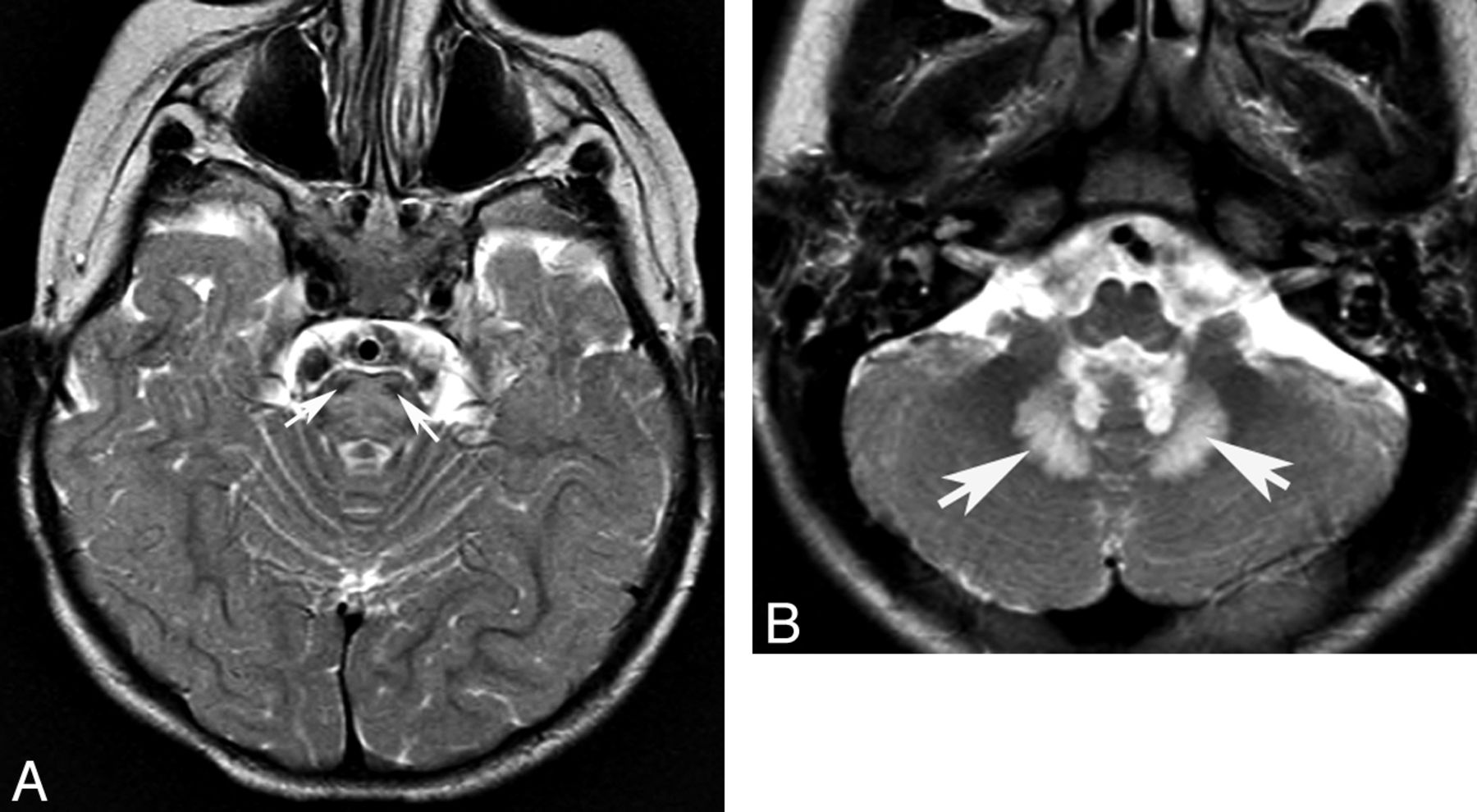
Complex II mitochondriopathy. Axial T2 images demonstrate hyperintense signal in the pons. (A, note sparing of corticospinal tracts, white arrowheads) and hilum of the dentate nucleus (B).
DWI and MR spectroscopy are occasionally helpful in making a diagnosis. Most commonly, brain stem and/or cerebellum lesions with reduced diffusivity in a child with ataxia suggest an acquired (usually vascular, inflammatory, or toxic) cause. These considerations should be quickly assessed, and if they are deemed unlikely or are excluded on clinical grounds, an acute metabolic exacerbation from mitochondrial disorders, organic acidurias, and urea cycle disorders (Fig 10) can be considered. Some degree of urgency is required in these cases because reduced diffusivity in a child with ataxia represents an active and sometimes treatable condition. MR spectroscopy is less often helpful. Reduced NAA is a common finding but is completely nonspecific. Elevated lactate can be observed in patients with mitochondrial disease or infantile neuronal ceroid lipofuscinosis (Fig 10).
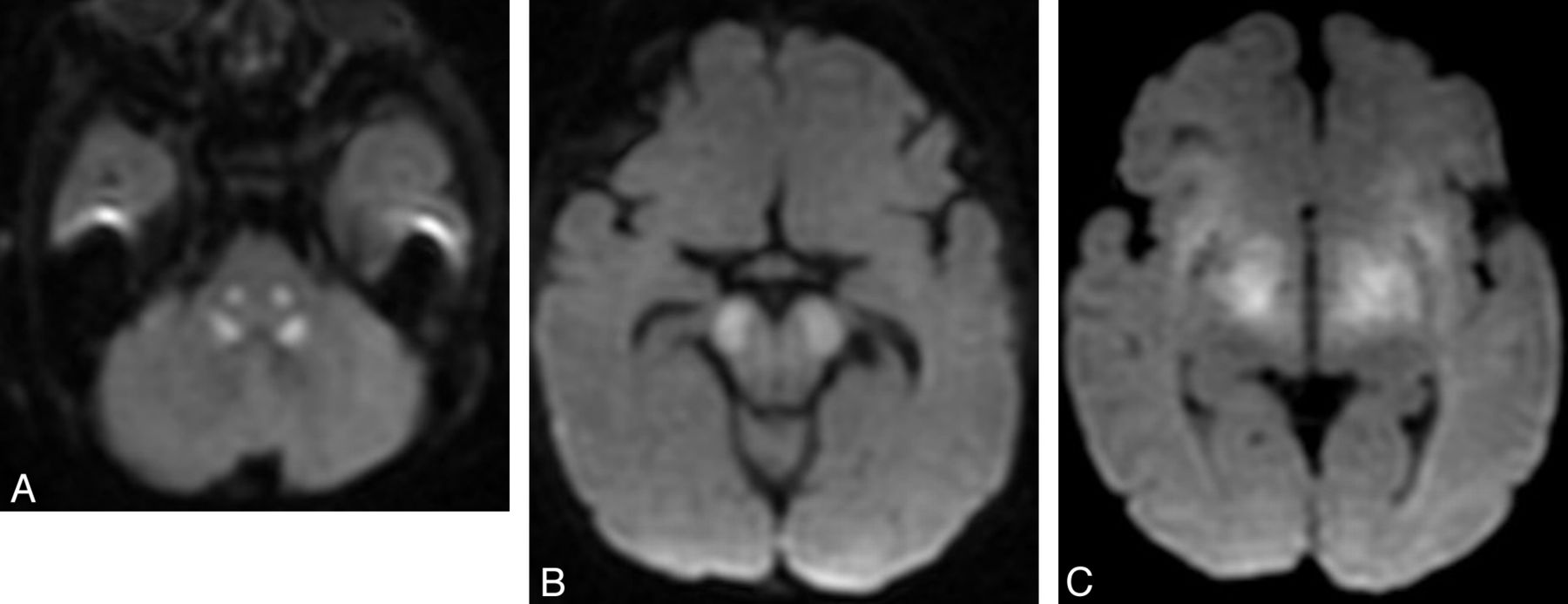
Diffusivity changes in mitochondrial disorder. Axial DWI images show brain stem reduced diffusion in the central tegmental tracts (A), cerebral peduncles (B), and subthalamic regions. Single-voxel proton MR spectroscopy (C) from the same patient shows reduced NAA and markedly elevated lactate (doublet at 1.33 cpm).
Calcification is a rare finding in CAC, and most commonly is associated with endocrine dysfunction or infection. However, if an acquired process is excluded by clinical and laboratory examinations, mitochondrial diseases, Cockayne syndrome, biotinidase deficiency, cerebrotendinous xanthomatosis, and carbonic anhydrase deficiency should be considered as potential causes.
Malformative Pattern of CAC
Although not all patients with cerebellar malformations have ataxia, patients with ataxia and the malformative pattern usually present during the neonatal or early infantile period with static ataxia and associated motor disability, muscular hypotonia, or impaired development (On-line Table 2). If the malformation involves supratentorial structures, other manifestations may predominate and the ataxia may not be detected. Although no treatment is available, a specific diagnosis is important for accurate genetic counseling, understanding, and classification of these disorders. Typical MR imaging findings in these patients are abnormal size or morphology of the brain stem or cerebellum. High-resolution thin-section 3D T1- and T2-weighted sequences with multiplanar reconstructions are most useful for the diagnosis of these abnormalities. In addition, DTI can be useful to identify axonal pathways, which may be anomalous.
One useful approach to posterior fossa malformations is to initially divide the cerebellar malformations into those with hypoplasia and those with dysplasia.22 Hypoplasia indicates a small cerebellum that has fissures of normal size compared with the folia, whereas dysplasia signifies an abnormal folial pattern or the presence of heterotopic nodules of gray matter. After this division, malformation can be further classified as focal (localized to either a single hemisphere or the vermis) or generalized (involved both cerebellar hemispheres and the vermis). Brain stem anomalies may be associated with hypoplasia or dysplasia of the cerebellum, though isolated brain stem anomalies can also be associated with ataxia in children.
Localized cerebellar hypoplasia is usually associated with a cerebellar cleft or an irregular cortex and is most commonly caused by a prenatal disruption of the cerebellar development, usually by infarct or hemorrhage (Fig 11). Rarely, localized hypoplasia may be genetic with localized mosaicism. In children with CAC, focal hypoplasia is usually restricted to the vermis. Mild-to-moderate vermis hypoplasia is a well-recognized finding in many developmental disorders, and the literature describes a large number of diseases with this pattern.6,10,34⇓–36
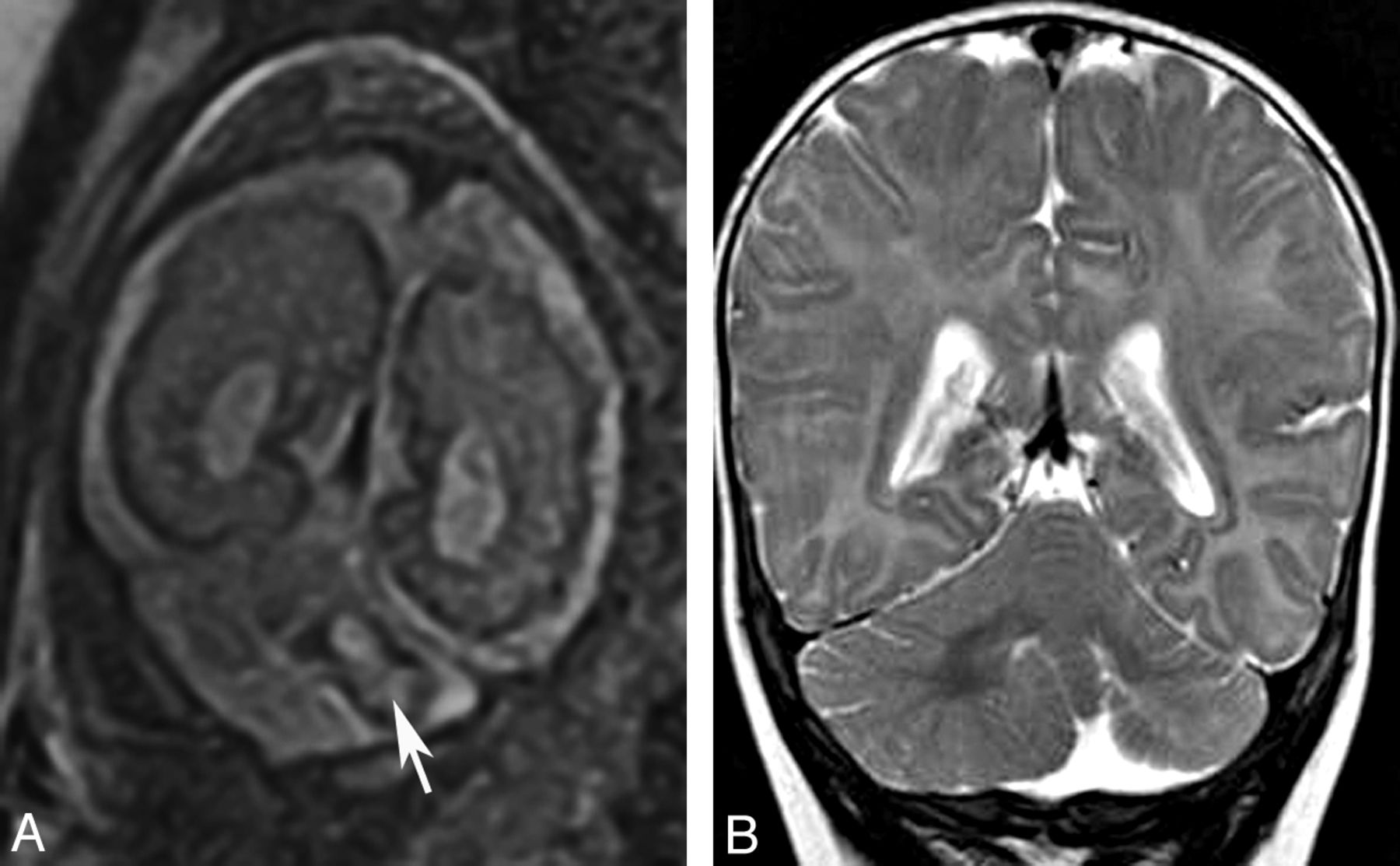
Localized cerebellar hypoplasia. Fetal coronal T2 image (A) shows a left-sided cerebellar hematoma (arrow). Postnatal coronal T2 image (B) demonstrates hypoplasia of the left cerebellar hemisphere.
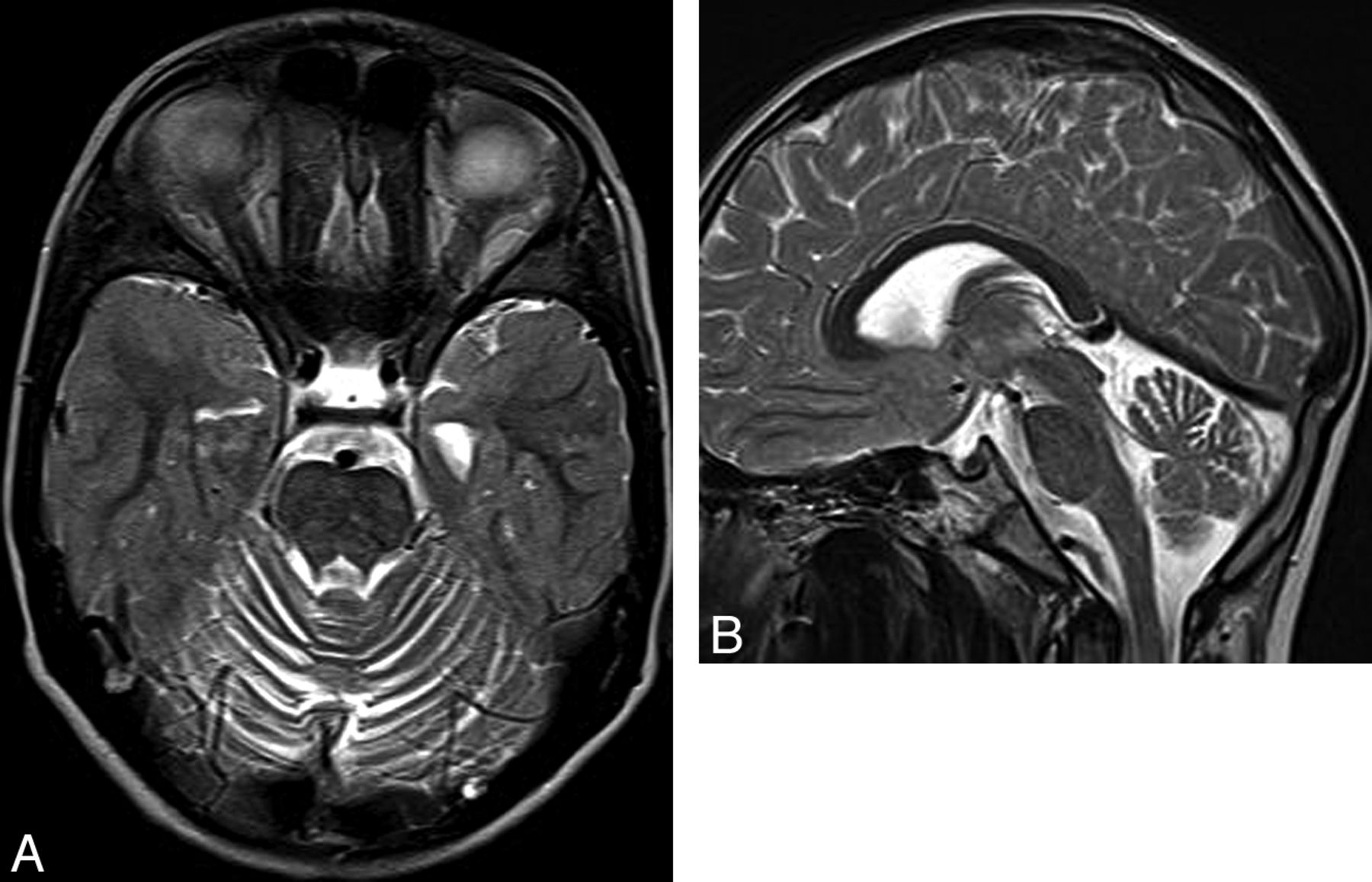
Severe generalized cerebellar hypoplasia is a common MR imaging feature in pediatric neuroimaging.20,35,37,38 (As a general rule about size, the normal cerebellar vermis extends from the inferior colliculi to the obex on the midline sagittal MR image of a neonate and from the intercollicular sulcus to the obex in an older infant or child, Fig 12). As discussed earlier, cerebellar hypoplasia is associated with pontine hypoplasia if growth arrest begins prenatally. Cerebellar hypoplasia has been described in the context of a wide variety of disorders, including hindbrain malformations, malformations of cortical development, extremely premature births (<1500 g or <30th week of gestation), chromosomal abnormalities (eg, trisomy 13, 18, or 21), and in utero exposure to toxins or infectious agents (eg, cytomegalovirus, On-line Table 2). Most interesting, it can also be caused by metabolic disorders, presumably because some of the same genes and protein products that are important in maintaining metabolic homeostasis after birth are important in guiding normal development in utero.

Cerebellar hypoplasia. Sagittal T1 image of a neonate shows severe brain stem and vermis hypoplasia. The superior vermis is below the intercollicular sulcus and above the obex. Note severe supratentorial atrophy, suggesting prenatal onset of volume loss.
Another group of disorders that presents with CAC and displays a small cerebellum and pons at the time of initial imaging is congenital disorders of glycosylation, particularly type 1a. Congenital disorders of glycosylation is a group of increasingly recognized diseases resulting from defects in the metabolism of glycoproteins. Congenital disorders of glycosylation type Ia (formerly known as carbohydrate deficient glycoprotein syndrome) is the most common form and has a variable clinical expression. Older children sometimes present with ataxia, mental retardation, and peripheral neuropathy. Neuroimaging shows signs of both cerebellar hypoplasia and atrophy, indicating that cerebellar damage likely begins in utero (Fig 13). Pontine hypoplasia and brain atrophy are usually associated findings.13

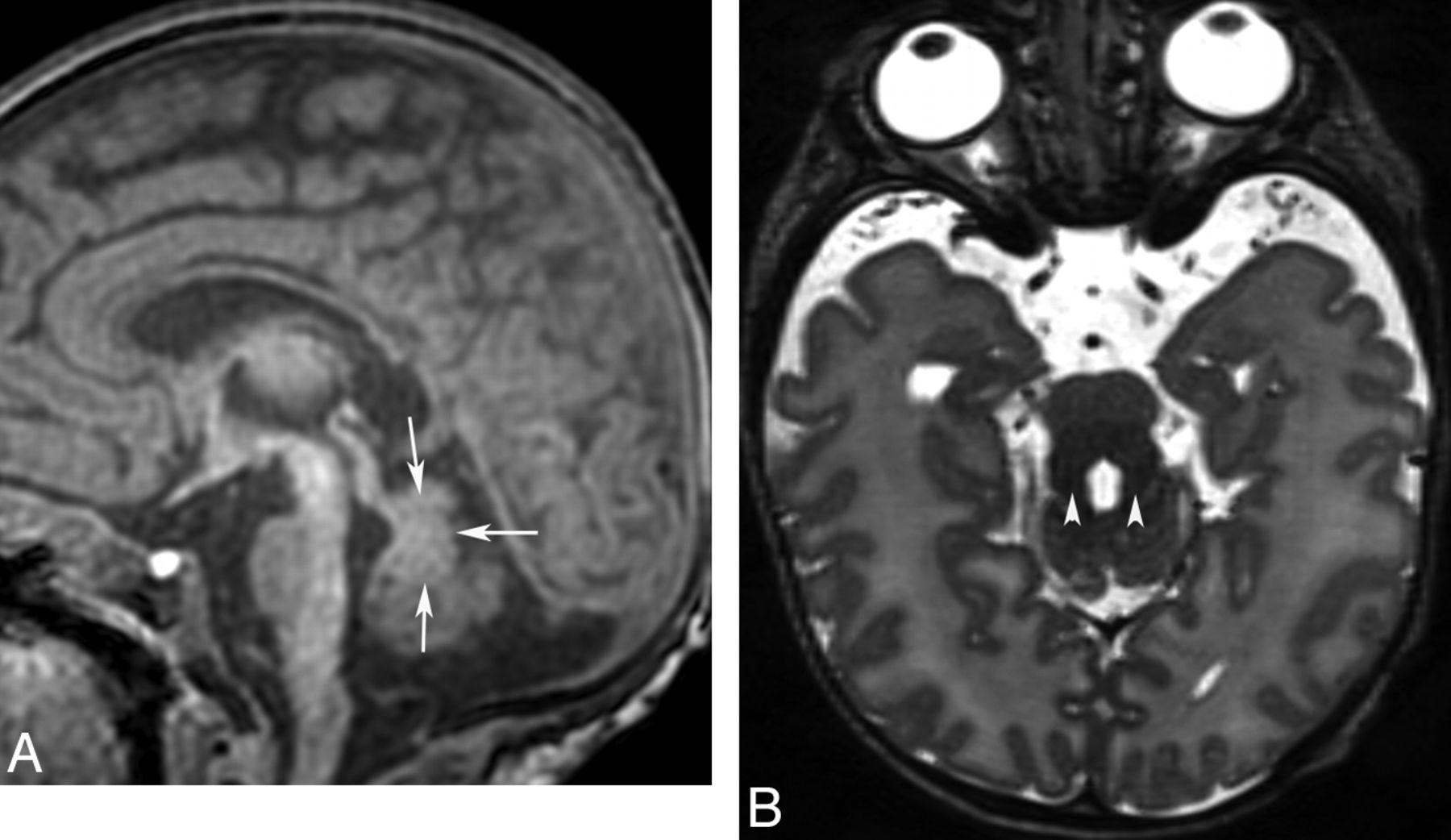
Congenital disorder of glycosylation (type Ia). Coronal T2 image shows a small cerebellum with very small cerebellar folia and enlarged fissures. This pattern probably reflects a process that begins in utero (hypoplasia) and progresses in infancy (atrophy).
Regarding the dysplastic type of isolated hindbrain malformation, the best characterized forms associated with focal dysplasia of the vermis are molar tooth–type malformation caused by mutations of primary ciliary protein genes (JSRD) and rhomboencephalosynapsis. “JSRD” is a term used to describe a number of conditions that have vermis hypoplasia and the distinctive “molar tooth” sign on MR imaging (Fig 14).16,18,39,40 It is considered a ciliopathy because most disorders of patients with this MR imaging sign seem to be associated with abnormal function of the primary cilia, specialized membrane-bound structures that project from the neuron and ependyma surface and serve many functions in development of the brain, kidneys, and eyes.39,40 In addition to the small dysmorphic vermis, imaging shows a small (sometimes dysmorphic) midbrain; thin isthmus (midbrain-pons junction); enlarged horizontal nondecussating superior cerebellar peduncles (molar tooth on axial images); and a triangular-shaped IV ventricle (“bat wing” on axial images).40

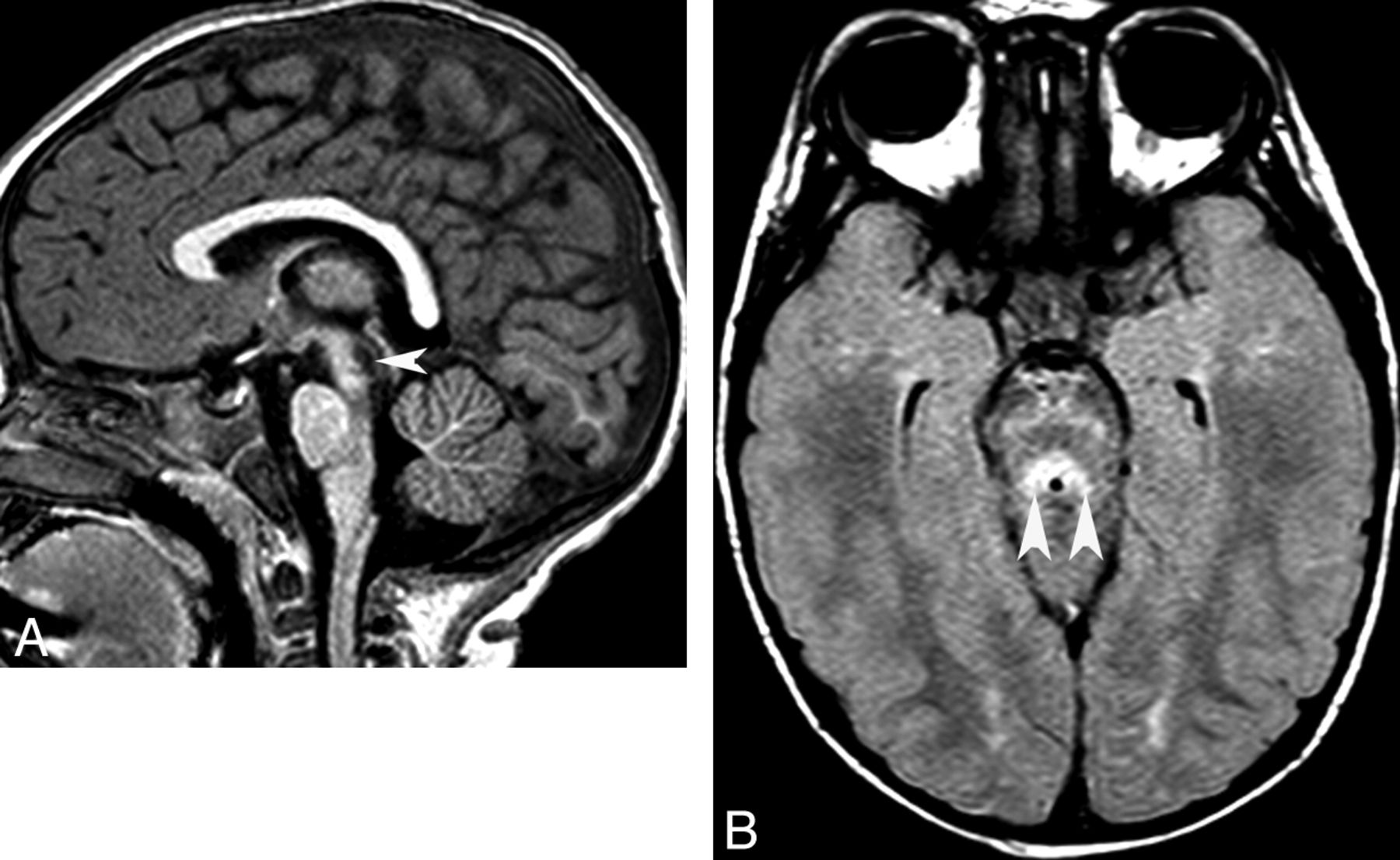
Joubert syndrome and related disorders (ciliopathies). Sagittal T1 (A) and axial T2 (B) images show a small dysgenetic vermis (arrows) and large horizontal superior cerebellar peduncles (arrowheads, molar tooth sign).
Although the pathogenesis is still controversial, rhomboencephalosynapsis has been postulated to be the result of a dorsoventral patterning defect in the caudal-mesencephalon and rostral-most rhombencephalon, leading to a lack of induction of the cerebellar vermis.18,41 It is characterized by a midline continuity of the infratentorial structures such as the deep cerebellar nuclei, superior cerebellar peduncles, and cerebellar hemispheres. Additional findings may include hydrocephalus (due to aqueductal stenosis, probably the most common finding outside the cerebellum), fusion of the midbrain colliculi, callosal agenesis, and absence of the septum pellucidum (Fig 15); these likely reflect a defective dorsoventral patterning disruption in the mesencephalon and caudal diencephalon.

Rhomboencephalosynapsis. Axial T2 (A) and coronal T2 (B) images show continuity of the deep cerebellar white matter across the midline due to complete absence of the vermis.
Other inherited diseases that cause nonprogressive ataxia with mild-to-moderate vermis hypoplasia include OPHN1 and Opitz/GBBB syndromes.6 Although the phenotype is variable, people with the OPHN1 syndrome usually present with impaired cognition and imaging showing vermis hypoplasia. Screening for OPHN1 mutations in a large cohort of boys with cognitive impairment and vermis hypoplasia led to identification of mutations of this gene in 12%.42 Patients with Opitz/BBB syndrome are also characterized by developmental delay, ataxia, and midline abnormalities, including hypertelorism, cleft lip/palate, esophagolaryngotracheal defects, imperforate anus, and hypospadias. Brain MRI consistently shows anterior vermis hypoplasia. Distinguishing genetically heritable syndromes from isolated cases of vermian/cerebellar hypoplasia is important for both prognosis and genetic counseling.
A number of reports have described patients with ataxia in whom imaging revealed cerebellar cortical dysgenesis, characterized by abnormal folia orientation and abnormal cortical thickness and resulting hypertrophic and/or hypoplastic cerebellar hemispheres.18,22,43 The cause of these malformations is not known, but they are usually associated with supratentorial anomalies (Fig 16) and are believed to be nongenetic (disruptive), possibly caused by infection or vascular causes.

Cerebellar cortical dysgenesis. Axial T1 images (A and B) and a coronal T1 image (C) demonstrate abnormal cerebellar foliation. The cause of this is usually unknown. Hydrocephalus is also present (C).
The Dandy-Walker malformation is not a common cause of ataxia in children. Although severe hypoplasia of the vermis and cerebellar hemispheres is a classic finding of this very heterogeneous malformation, the presence of vermis hypoplasia or a large cisterna magna does not establish a diagnosis of Dandy-Walker malformation. It is very likely that Dandy-Walker malformation is currently overdiagnosed, and the diagnosis should be made with caution, especially when detected on fetal sonography or MR imaging.18 The cerebellum grows late compared with the cerebrum and often looks small on fetal imaging. Comparison of cerebellar size with normative age-matched measurements is critical.
Many malformations of cortical development are associated with cerebellar dysgenesis, including lissencephalies (particularly those caused by mutations of the Reelin pathway or TUBA1A), cobblestone malformations, and some types of polymicrogyria.18 When present, CAC is not a major clinical feature of cerebral malformations of cortical development. However, ataxia has been described in patients with merosin-positive congenital muscular dystrophy and evidence of cerebellar hypoplasia on MR imaging.44 This association it is not surprising because the general mechanisms regulating cortical formation in the cerebrum are shared by other regions of the central nervous system, including the cerebellum. Mutation of the very low-density lipoprotein receptor gene, which functions within the Reelin pathway (together with RELN, ApoER2, and DAB1) is the cause of an autosomal recessive syndrome of nonprogressive cerebellar ataxia and mental retardation known as the disequilibrium syndrome.36,45 Imaging shows significant cerebellar hypoplasia and mild cerebral cortex thickening and gyral simplification. Therefore, the supratentorial cortex should be routinely evaluated in children with CAC because the supratentorial anomalies may be more specific than those in the posterior fossa.
Finally, malformations of the brain stem are a growing group of anomalies of the CNS that have been recently classified.16 Considering the common embryologic pathways involving both the pons and cerebellum and the evidence of intense axonal connections between these, it is likely that primary brain stem abnormalities can cause CAC.16,18 Indeed, although brain stem anomalies typically cause cranial neuropathies, ataxia has been described in patients with pontine tegmental cap dysplasia and pontine dysgenesis with clefts. MR imaging shows a small ventral pons with a round or beaklike dorsal pontine protuberance (tegmental cap) in the former (Fig 17)46 and dorsal or ventral clefts in the pons in the latter. In both disorders, color fractional anisotropy maps from DTI show abnormal configuration of the WM tracts in the brain stem.

Pontine tegmental cap dysplasia. Sagittal T2 image (A) shows a rounded “cap” over the dorsal pons (arrow). Axial T1 image (B) demonstrates a small pons and a flat anterior border of the forth ventricle secondary to transversely oriented axons.
Conclusions
Neuroradiologists should be aware that ataxia in children is a common manifestation of many disorders. Some can be suggested or diagnosed by imaging; others can be prevented by genetic counseling or treated if detected in early stages. Diagnosing these disorders requires a careful analysis of posterior fossa structures and knowledge of the disturbances of normal anatomy caused by these disorders. This review, hopefully, clarifies the causes of inherited CAC and adds to the knowledge of many of these diseases.
Indicates open access to non-subscribers at www.ajnr.org
References
- © 2013 by American Journal of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.