
Abstract
SUMMARY: HGPS is a rare syndrome of segmental premature aging. Our goal was to expand the scope of structural bone and soft-tissue craniofacial abnormalities in HGPS through CT or MR imaging. Using The Progeria Research Foundation Medical and Research Database, 98 imaging studies on 25 patients, birth to 14.1 years of age, were comprehensively reviewed. Eight newly identified abnormalities involving the calvaria, skull base, and soft tissues of the face and orbits were present with prevalences between 43% and 100%. These included J-shaped sellas, a mottled appearance and increased vascular markings of the calvaria, abnormally configured mandibular condyles, hypoplastic articular eminences, small zygomatic arches, prominent parotid glands, and optic nerve kinking. This expanded craniofacial characterization helps link disease features and improves our ability to evaluate how underlying genetic and cellular abnormalities culminate in a disease phenotype.
ABBREVIATIONS:
- CDC
- Centers for Disease Control and Prevention
- HGPS
- Hutchinson-Gilford progeria syndrome
- OFC
- occipitofrontal circumference
- TMJ
- temporomandibular joint
- WHO
- World Health Organization
HGPS is a rare syndrome of segmental premature aging.1 HGPS results from a sporadic autosomal dominant mutation of the LMNA gene, which encodes for the inner nuclear membrane protein lamin A.2,3 Progerin, the disease-causing aberrant lamin A protein, causes alterations in the nuclear structure,4 cellular life span,5 and transcription.6 Progerin is expressed in all differentiated cells, supporting a multisystem disease phenotype in HGPS.7
Children with HGPS typically appear healthy at birth and within the first year of life develop severe failure to thrive with sclerodermatous skin changes.8,9 Classic craniofacial features develop with age and underlying disease culminates in death from myocardial infarction or stroke at an average age of 13 years.8,10
Close examination of common and disparate findings in HGPS and aging may inform both fields of study. Clinical features that resemble premature aging include alopecia, joint contractures, progressive mandibular resorption, low bone density, lipoatrophy with limb wasting, and global atherosclerosis.8,11 Age-related conditions such as Alzheimer disease, dementia, osteoarthritis, and cancer are absent. Cognitive function and motor development are normal. In addition, there is growth failure, skeletal dysplasia, and lack of pubertal development.12
Prior reports of craniofacial features are based on plain film findings and autopsy results.10,13,14 In this study of a large cohort of patients with HGPS, we reviewed CT, MR imaging, and radiographs to quantify previously noted and new craniofacial imaging features to expand our understanding of the natural history of the disease.
Materials and Methods
Patients and Data
Patients were identified from The Progeria Research Foundation Medical and Research Database (Brown University Center for Gerontology and Healthcare Research, Providence, Rhode Island). Eligible individuals had a genetic and/or clinical diagnosis of HGPS and at least 1 MR or CT of the head or neck available for review. The diagnosis of HGPS was genetically determined by either the classic exon 11 (c. 1824 C>T) or a nonclassic progerin-producing LMNA mutation within exon 11 or the exon/intron border, plus the typical clinical phenotype.15
Data abstraction included indications for neuroimaging studies, OFC, and medications. Height-age was determined by using the CDC and WHO child growth standards for boys and girls.16,17 Abstracted OFC measurements from patient charts were expressed as percentiles by height-age.
Neuroimaging Studies
A neuroradiologist (V.M.S.) and a neurologist (N.J.U.) interpreted all neuroimaging studies, including MR; CT; and radiographs of the face, neck, and skull, together in consensus-reading sessions. All imaging studies were reviewed twice. The purpose of the first review was to catalog known and novel bony and soft-tissue craniofacial features by using visual/subjective assessment. The purpose of the second review was to establish the presence or absence of each individual imaging feature. Only the second review was used to determine prevalence.
This retrospective Health Insurance Portability and Accountability Act–compliant study was approved by the institutional review boards of Brown University and Rhode Island Hospital with informed consent from parents or legal guardians.
Results
Demographic Features
Patient characteristics and types of neuroimaging studies assessed are presented in Table 1. Twenty-five children provided a total of 98 neuroimaging studies from birth to 14.1 years of age (mean, 6.5 years). There were 11 females and 14 males. Eighteen children were diagnosed with classic HGPS, 3 patients had the nonclassic phenotype and nonclassic progerin-producing mutations based on confirmed LMNA mutational analysis, and 4 patients were diagnosed on the basis of phenotypic disease expression.
Patient characteristics
Neuroimaging Results
The most frequent indications for imaging were evaluation of acute neurologic symptoms and/or diagnostic evaluation or follow-up of a prior imaging finding (Table 2). Twenty disease-related abnormalities were detected, including previously described craniofacial features (Fig 1) and 8 newly identified features. Disease-related features were categorized into abnormalities of the scalp, calvaria and skull base, facial bone, soft tissue, and orbits (Table 3). Frequency is expressed as the number of patients with a positive finding relative to the number of patients in whom imaging allowed adequate assessment.
Indications for imaging
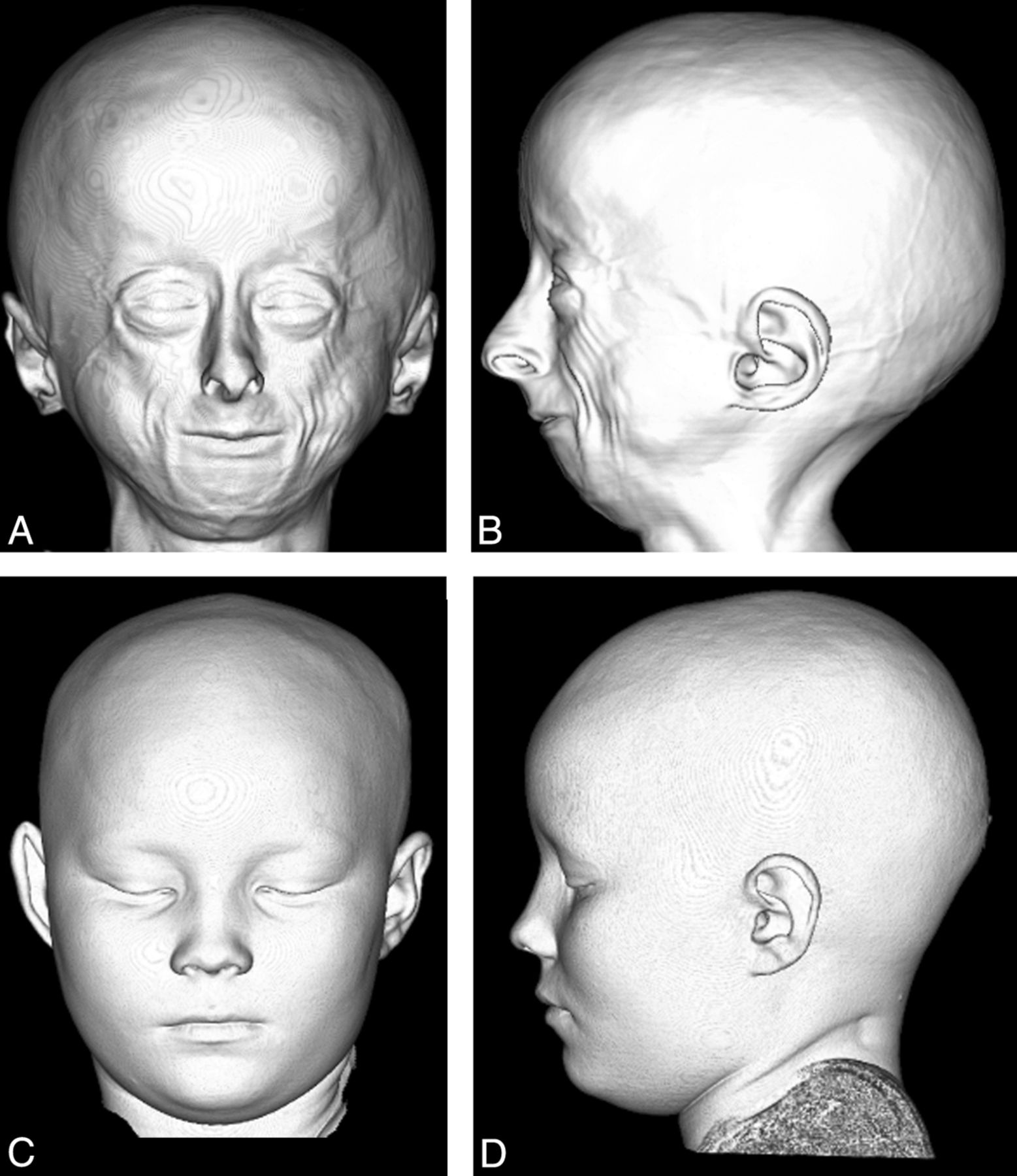
Anterior (A) and lateral (B) projection of a 3D CT shaded-surface display of the head demonstrates craniofacial disproportion, prominent eyes, hypotelorism, narrow nasal bridge with broad tipped nose, small face and mandible, lack of facial fat, and prominent veins in a 9-year-old child with HGPS compared with an age-matched control (C and D).
Anatomic abnormalities of head and neck in HGPS
Scalp, Calvarial, and Skull Base Features
Thinning of the calvarium was seen in 20/21 (95%) individuals and was accompanied by a paucity of scalp fat in 21/23 (91%) (Fig 2). A mottled appearance of the skull was seen in 10/17 (59%) patients (Fig 3), most commonly in the frontal, parietal, and sphenoid regions. No overt bony destruction or osteolysis of the calvaria was seen. Two individuals had skull fractures. Prominent vascular markings of the bony calvaria were observed in 9/10 (90%) (mean age, 6.8 years; range, 0–13.1 years) (Fig 3), including 3 children younger than 1 year of age.

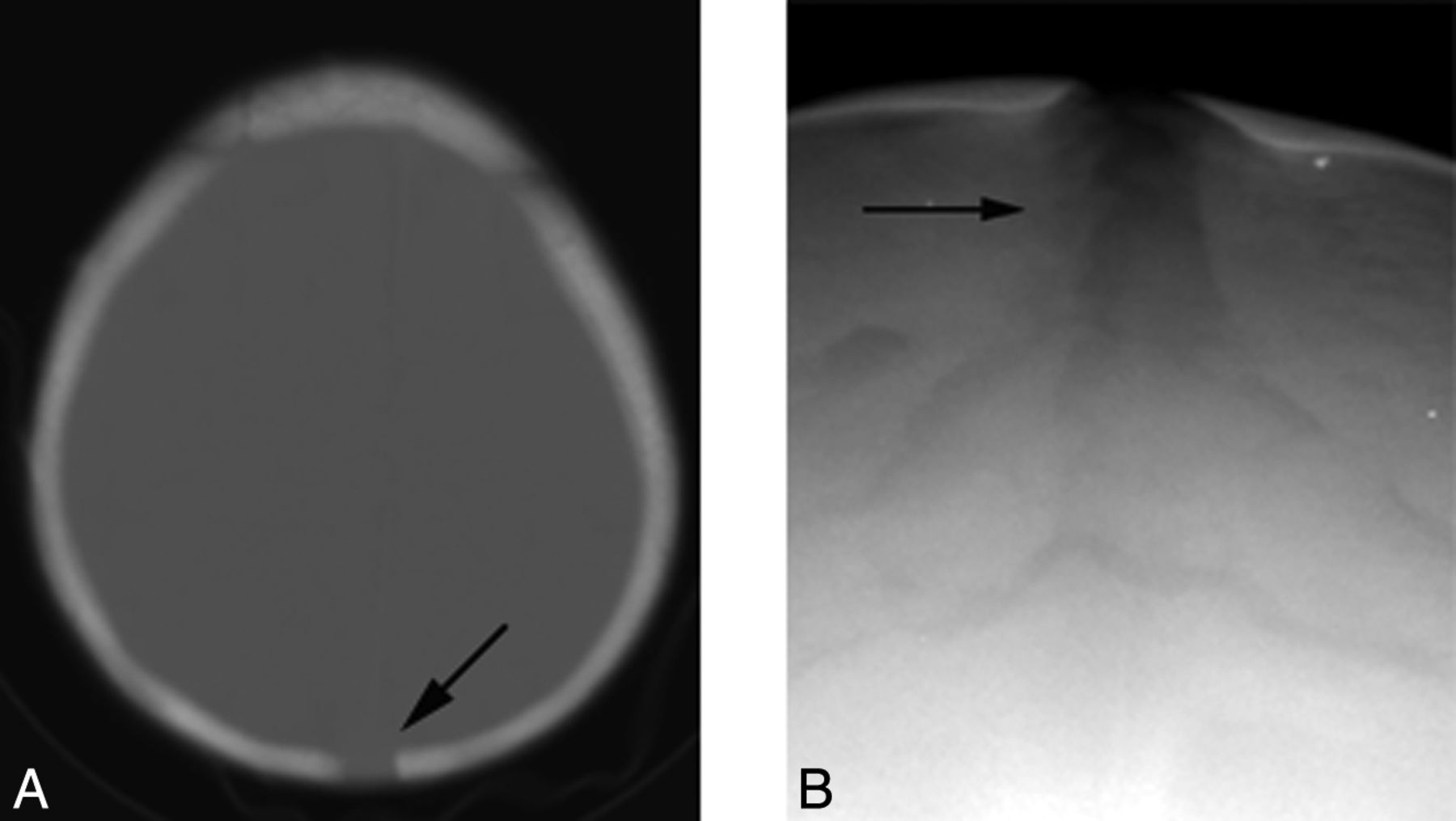
Sagittal CT reformatted image of a 9-year-old child with HGPS (A) demonstrates a thin calvarium, paucity of scalp fat, a J-shaped sella (single arrow), and a patent anterior fontanel (double arrow) compared with the control (B).

A 10-year-old child with HGPS. Axial CT image reveals a mottled appearance of the calvarium (A) and prominent vascular markings (arrow, B).
In 96% of normally developing children, closure of the anterior and posterior fontanels occurs by 2 years and 2 months of age, respectively.18,19 If one excludes children younger than 2 years of age, the anterior fontanel remained patent in 9/16 (56%) (range, 2.1–12.1 years; mean, 7.4 years) (Fig 2). Adequate visualization of the posterior fontanels was limited but was clearly open in 3 children, 2.1, 3.5, and 9.4 years of age. Widened sagittal, squamous, and lambdoid sutures were observed in 7/17 (41%) children (Fig 4).

A 2-year-old child with a persistent patent posterior fontanel (arrow, A) on axial CT and a widened sagittal suture (arrow, B) on the anteroposterior radiograph of the skull.
A large cranium relative to the facial size was seen in 23/25 (92%) (Fig 1). Average patient length/height for age was well below the third percentile, and average patient length/height for age was 50% of chronologic age. By contrast, the mean head circumference normalized to height-age was at the 56th percentile (n = 254 observations from all 25 children, range fifth to 97th percentile).
A J-shaped sella was observed in 17/19 (89%) children (Fig 5).
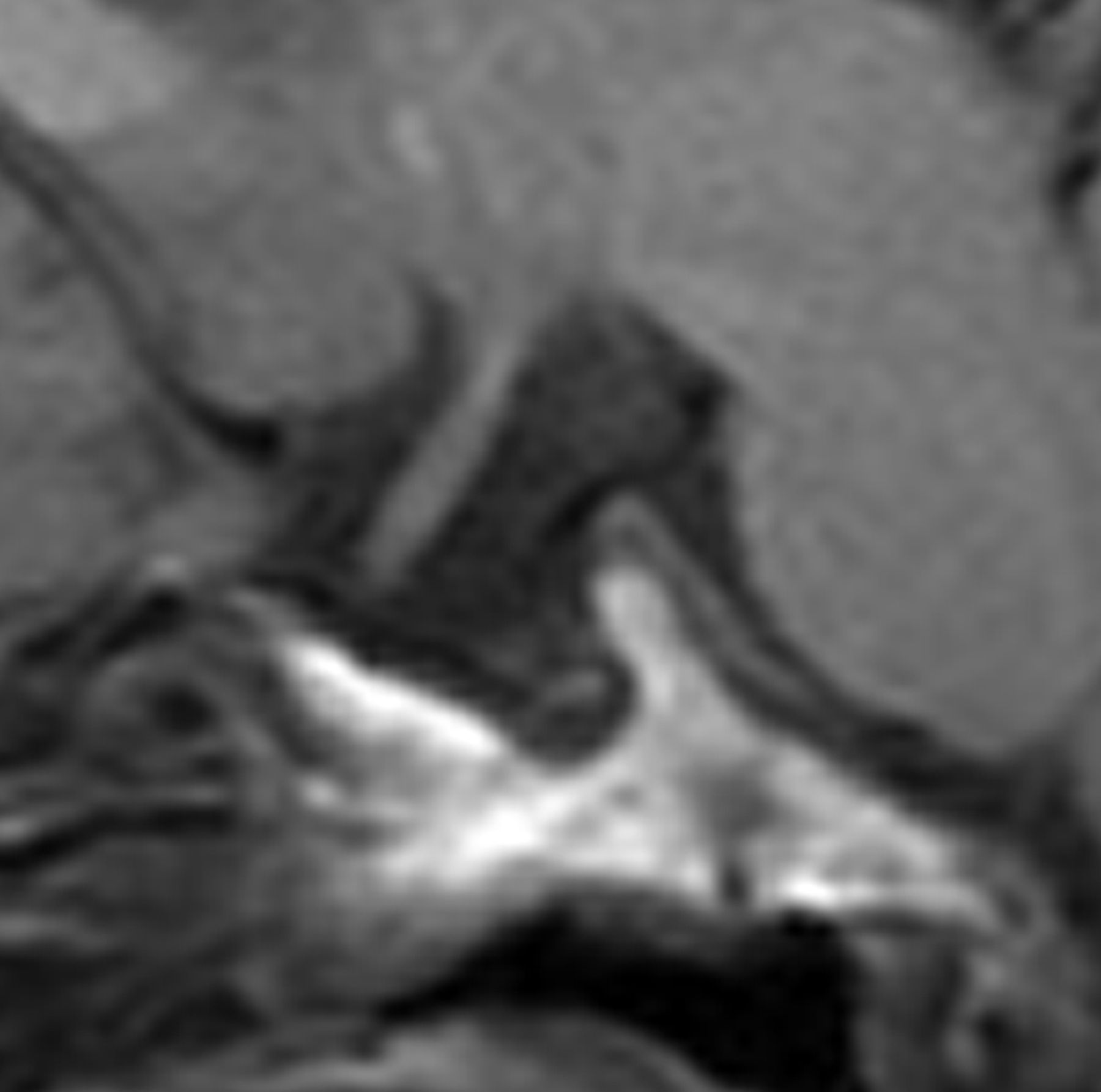
Sagittal T1-weighted MR image shows a J-shaped sella.
Oral Maxillary, Zygomatic Arch, and Parotid Gland Features
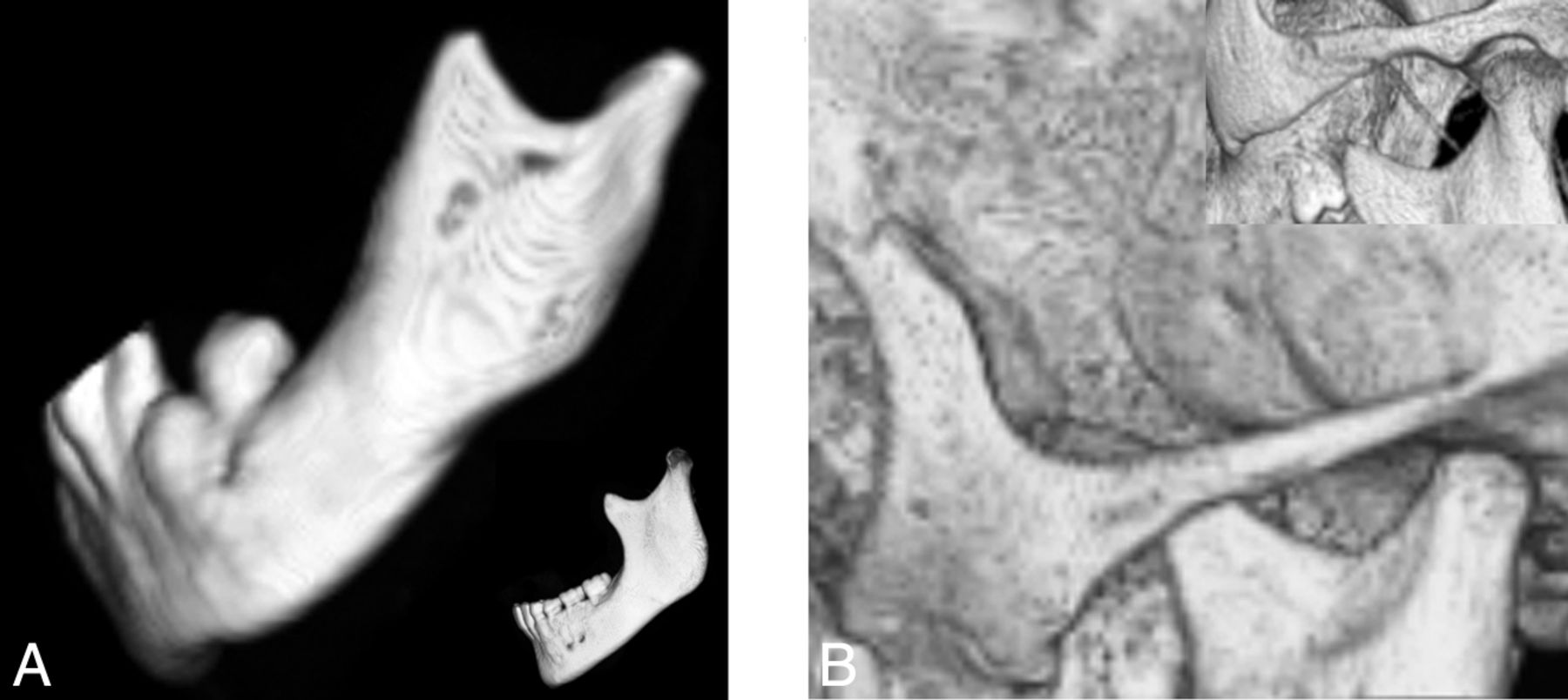
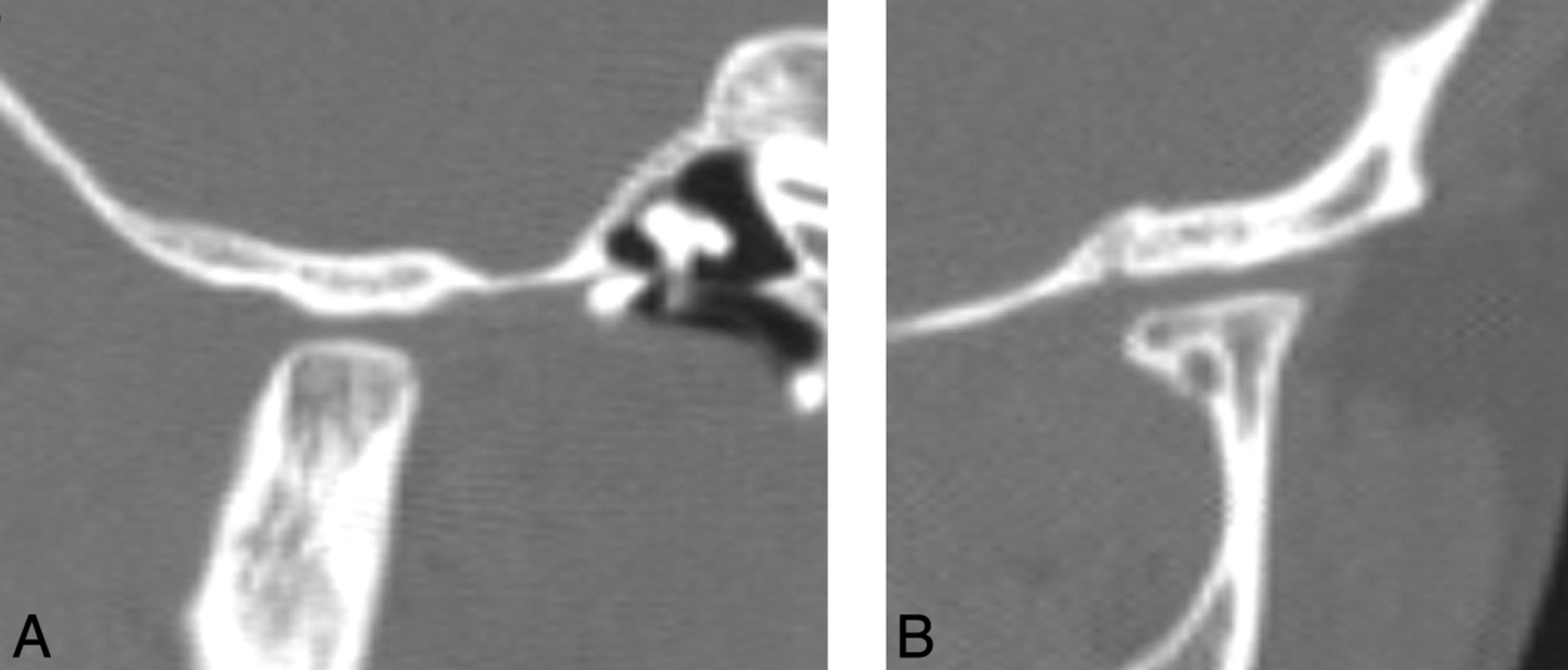
A short mandibular ramus was seen in 15/18 (83%) patients, with a gracile thin zygomatic arch in 6/12 (50%) (Fig 6). A shallow glenoid fossa with a hypoplastic or absent articular eminence and flattening of the mandibular condyle were seen in 6/14 (43%) (Fig 7). A V-shaped palate was seen in 9/20 (45%) children, and a disorganized dentition was seen in 10/20 (50%) (Fig 8).
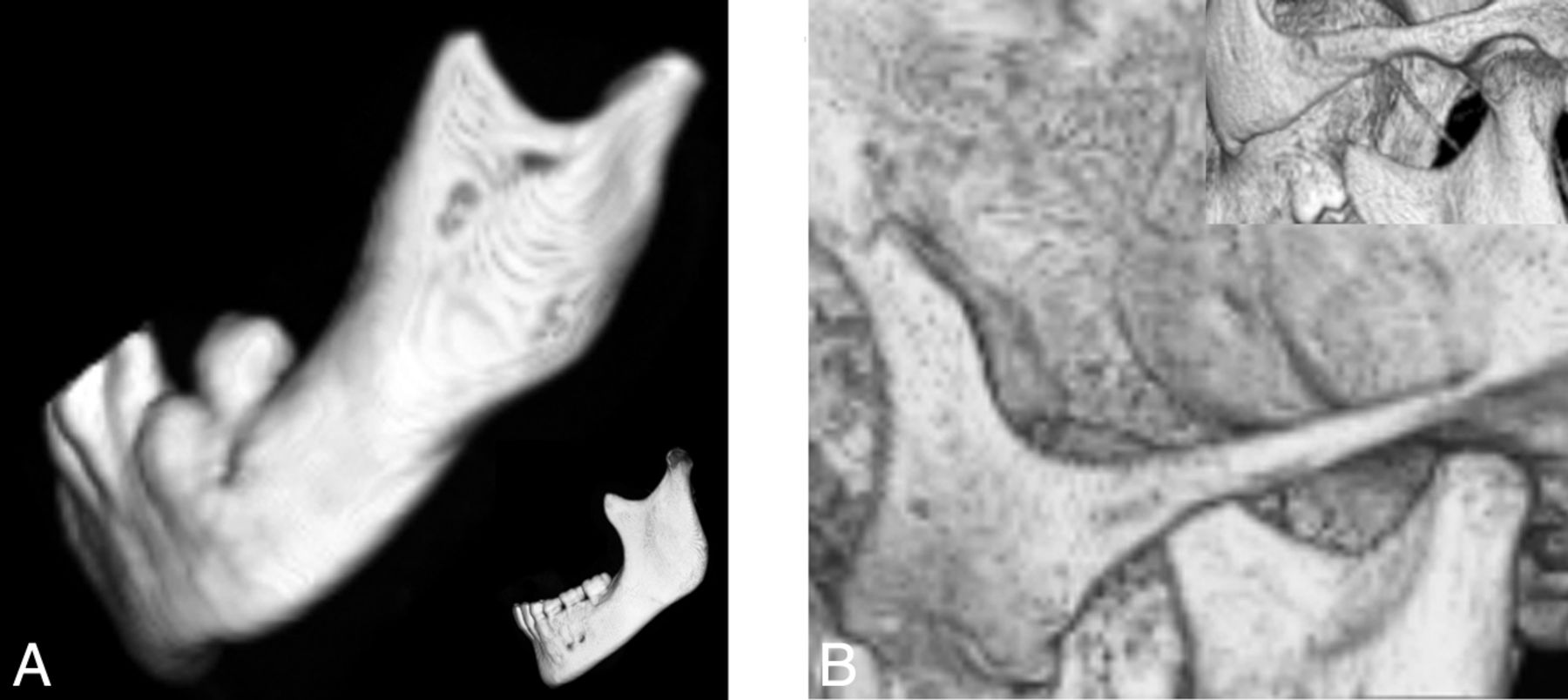
Lateral projection of a 3D CT shaded-surface display reveals a short mandibular ramus, a steep mandibular angle with disorganized dentition (A), and a thin gracile zygomatic arch in a child with HGPS (B) compared with a healthy age-matched control (insets).

A, Lateral CT reformatted image demonstrates flattening of the condylar head, a shallow glenoid fossa, and a hypoplastic articular eminence. B, Coronal CT reformatted image of a child with HGPS shows a flattened condylar head and a shallow glenoid fossa.

Axial T2-weighted MR imaging demonstrates a V-shaped palate, disorganized dentition, and prominent parotid glands in child with HGPS (A) compared with the control (B).
Prominence of the parotid glands was seen in 13/13 (100%) children (Fig 8). No intrinsic abnormality of the parotid gland was observed.
Orbital Features
Hypotelorism was noted in 19/22 (86%) children (Fig 9). Kinking of the optic nerves was seen in 17/19 (89%) (Fig 10). We did not detect any patients with exophthalmos, exorbitism, or overabundance of retrobulbar fat on imaging. No abnormality was seen in the extraocular muscles or globes.

Anterior projection of a 3D CT shaded surface display of the head in a child with HGPS (A) shows hypotelorism, small mid- and lower face, and a disorganized dentition compared with the control (B).
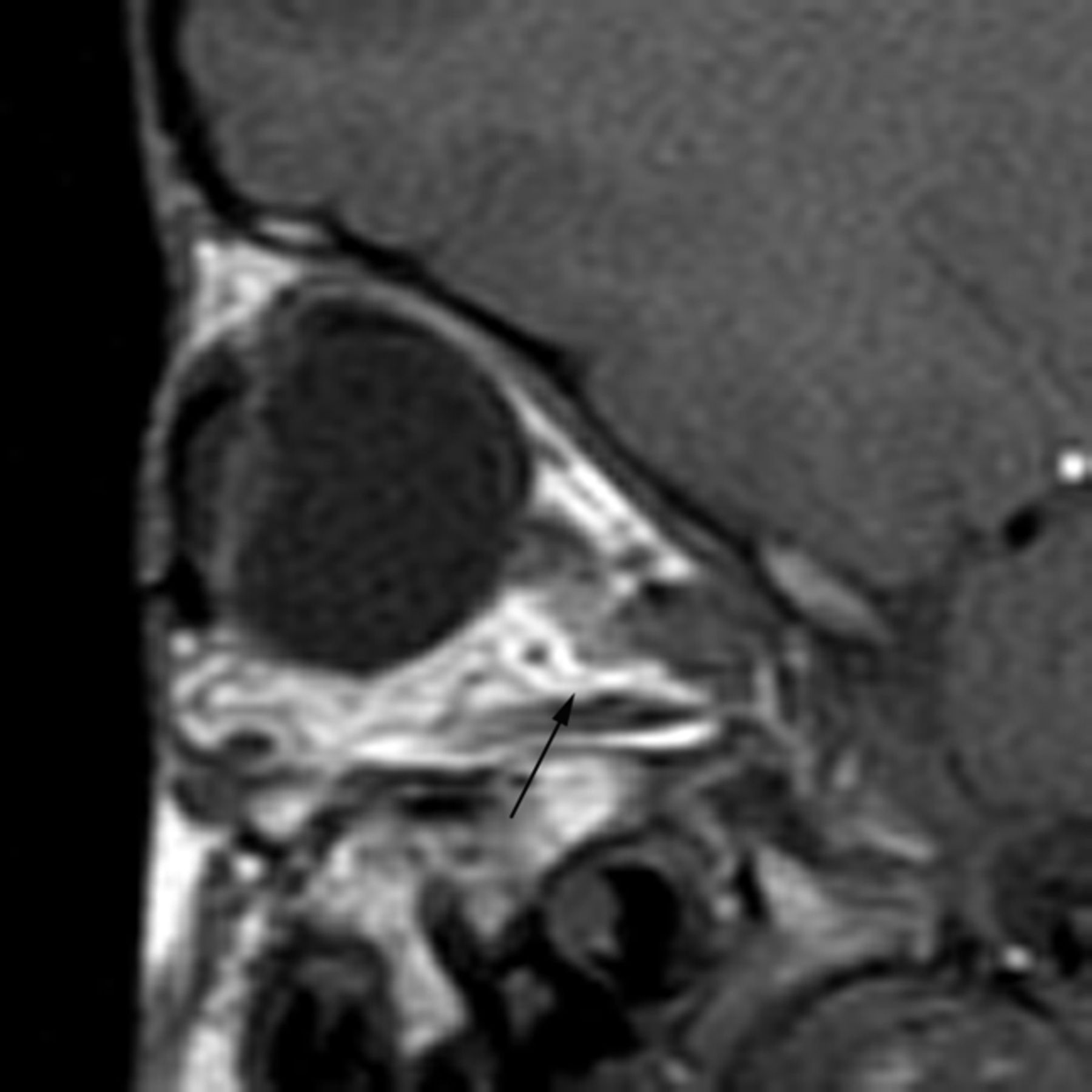
Sagittal T1-weighted MR image demonstrates kinking of the optic nerve (arrow).
Discussion
We established a prevalence for 20 different disease-related head and neck abnormalities in this cohort; of these, 8 have not been previously reported, to our knowledge. The morphologic spectrum of craniofacial features in HGPS is considerably expanded. Both new and previously identified features occurred with high frequency (41%–100%), which reinforces the consistency of pathobiology among patients.
Calvarial and Skull Base Changes
We observed bony abnormalities of the calvarium and skull base, including thinning and mottling of the calvarium, prominent vascular markings, delayed closure of the anterior and posterior fontanels, widening of calvarial sutures, and a J-shaped sella.
We did not observe bony destruction of either the calvaria or skull base to suggest the presence of osteolysis, a feature frequently observed in the clavicle, digits, and ribs. However, we noted mottling of the flat bones of the skull and postulate that this finding may represent bony demineralization, similar to the juxta-articular demineralization seen on radiographs.10,14 The osteopenia of long bones is accompanied by abnormalities in structural rigidity and is thought to represent a skeletal dysplasia, rather than the typical osteoporosis of aging.12,20,21
Delayed closure of the anterior and posterior fontanels and widening of the calvarial sutures in HGPS have been reported.8 In our cohort, patients as old as 9 years exhibited persistently patent anterior and posterior fontanels. Widened calvarial sutures were also common. Bone growth at the calvarial sutures and fontanels occurs via intramembranous bone deposition at suture edges.22 Widened sutures and patent fontanels reflect a disturbance in the synchronized process of osteoblastic bone apposition and osteoclastic bone resorption in coordination with the growth of the underlying brain.23
We frequently observed a thin calvarium, which has been noted on autopsy specimens.9,10 The thinness of the skull bone may reflect delayed evolution of the normal bony remodeling of the skull to its mature organization, similar to stunted linear skeletal growth observed elsewhere. The normal infant skull thickens with age, transitioning from a unilaminar to a trilaminar composition. In the developing calvarium, appositional bone growth increases bone thickness.23 Disturbed appositional bone growth in HGPS has been suggested by the observation of laminations beneath the periosteum of long bones on radiographs.13
A predisposition toward skull fractures has been reported in HGPS.8 In our series, 2 children with skull fractures following trauma were noted. Fracture may result from the relative thinness of the calvarial bone in combination with osteopenia.
Prominent vascular markings of the calvaria were frequently observed, even in very young children. Generally, diploic veins are not visible before the third year of life and are not fully developed until 15 years of age. Meningeal vascular grooves along the inner table are not typically visualized until 12 years.24 The appearance of prominent vascular markings reflects either enlargement of nutrient canals within the diploic space or increased prominence of the vascular grooves along the inner table or both. Prominent diploic veins of the skull are frequently seen in the elderly. When decalcification or osteoporosis of the calvarium occurs, the vascular markings of the diploe stand out more prominently and the vascular grooves along the inner table appear more distinct.25 Therefore, calvarial demineralization may contribute to the aforementioned appearance as well. The prominent scalp veins seen clinically do not account for the bony remodeling because they are not pulsatile and are not bound by the periosteum along the outer table.
We frequently observed a J-shaped sella, which is an abnormal configuration of the sella turcica due to an exaggeration of the normal chiasmatic sulcus. This finding can be seen in genetic disorders, as an anatomic variant,26 and secondary to pathologic states such as chronic hydrocephalus. Most interesting, the only 2 patients in our series who did not have J-shaped sellas were also the only patients treated with recombinant growth hormone.
The appearance of macrocrania and craniofacial disproportion is a common clinical feature in HGPS. In our cohort, OFC measurements adjusted by height-age were normal; thus, it is likely due to the progressive remodeling of the facial bones rather than the presence of true macrocephaly.
Orbits
Clinically, children with HGPS have prominent eyes. Exophthalmos is not observed clinically and was not seen on imaging. We frequently observed optic nerve kinking and hypotelorism. Remodeling of the facial bones may result in a progressive foreshortening of the distance between the optic chiasm and the bony orbit, leading to optic nerve redundancy, further compounded by hypotelorism.
Mandible, Maxilla, TMJ, Zygomatic Arches, and Parotid Glands
Our findings confirm and expand on the factors affecting oral maxillary function.11,27 We observed temporomandibular abnormalities and thin gracile zygomatic arches, which have not been previously reported. A mixed dentition of the teeth of the mandible and maxilla has been described, giving rise to a “double row” appearance12 and reflects our common observation of a V-shaped palate and a disorganized dentition.
A small mandible-to-maxilla ratio with hypognathia was observed. Hypoplastic mandibles have been described, more prominently affecting the mandibular rami, steep mandibular angles, aberrantly positioned condylar processes, and loss of basal bone height.27 We identified short mandibular rami in combination with flattened mandibular condyles, shallow glenoid fossae, and hypoplastic or absent articular eminences. The flattened appearance of the condylar head bears a similarity to the abnormal remodeled metaphyses seen elsewhere in the appendicular skeleton in HGPS.9,12 Avascular necrosis, which is commonly observed in the femoral head in these children, could result in a flattened appearance of the mandibular condyle; however, the absence of jaw pain clinically makes this possibility less likely.
Most children with HGPS have a limited oral opening on clinical examination, and the aforementioned findings introduce possible etiologies. The limited oral opening may be due to a limited range of motion of the TMJ secondary to the abnormally configured mandibular condyle, glenoid fossa, and articular eminence. Periarticular fibrosis may be present, similar to the periarticular fibrosis seen elsewhere within the skeleton.11,28 Mandibular ankylosis is often noted in other syndromes associated with congenital and nonsyndromic mandbular hypoplasia.29 A foreshortened mandibular ramus will produce a smaller oral opening for any degree of rotation of the mandibular condyle within the glenoid fossa due to a smaller rotational arc. These factors may also contribute to the maintenance of vertical chewing seen in children with HGPS, who do not evolve to rotatory chewing, typical of normally developing children.11 Dedicated TMJ imaging could help elucidate these issues.
A “jowly” appearance of the face is often noted in HGPS. This is thought to be due to a relative loss of facial fat out of proportion to loss of buccal fat. Buccal fat, along with pubic fat, disappears relatively late in the course of the disease.8 We frequently observed prominent parotid glands, which could also account for the “jowly” appearance. It is unclear whether the prominence of the parotid glands is a result of true glandular hypertrophy or results from the progressively smaller relative dimensions of the facial bones along with decreasing facial fat.
Conclusions
This study constitutes the largest imaging review of craniofacial features in patients with HGPS, to our knowledge. We quantified many previously recognized craniofacial findings of HGPS and identified 8 new common features. We expanded the knowledge of the spectrum and prevalence of craniofacial features in HGPS and suggested potential etiologies and anatomic explanations for clinical issues of the head and neck. Slowing or arrest of these changes may provide new clinical-outcome parameters to follow progression, improvement, or delayed onset of findings with treatment interventions.
Acknowledgments
We thank Nalton Ferraro, MD, DMD, for his input on oral maxillofacial and mandibular function.
Footnotes
Disclosures: Leslie B. Gordon—RELATED: Grants: Rhode Island Hospital,* Brown University,* Support for Travel to Meetings for the Study or Other Purposes: The Progeria Research Foundation;* UNRELATED: Provision of Writing Assistance, Medicines, Equipment, or Administrative Support: Merck/Schering Plough Research Institute,* Comments: provide pharmacokinetics and trial medications at no cost. Patents: 2 patents pertaining to progeria (neither are paying entities), Travel/Accommodations/Meeting Expenses Unrelated to Activities Listed: The Progeria Research Foundation,* Other: volunteer medical director for the funding agency,* OTHER RELATIONSHIPS: Parent of a child with progeria. *Money paid to the institution.
References
- Received September 30, 2011.
- Accepted after revision November 18, 2011.
- © 2012 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}