
Abstract
SUMMARY: SNHL is a major cause of childhood disability worldwide, affecting 6 in 1000 children. For children with prelingual hearing loss, early diagnosis and treatment is critical to optimizing speech and language development, academic achievement, and social and emotional development. Cross-sectional imaging has come to play an important role in the evaluation of children with SNHL because otolaryngologists routinely order either CT or MR imaging to assess the anatomy of the inner ears, to identify causes of hearing loss, and to provide prognostic information related to potential treatments. In this article, which is the first in a 2-part series, we describe the basic clinical approach to imaging of children with SNHL, including the utility of CT and MR imaging of the temporal bones; we review the most recent proposed classification of inner ear malformations; and we discuss nonsyndromic congenital causes of childhood SNHL.
ABBREVIATIONS:
- ABR
- auditory brain stem response
- BCNC
- bony cochlear nerve canal
- CISS
- constructive interference in steady state
- CMV
- cytomegalovirus
- CN
- cochlear nerve
- CND
- cochlear nerve deficiency
- FN
- facial nerve
- FSE
- fast spin-echo
- IAC
- internal auditory canal
- IP-I
- incomplete partition type I
- IP-II
- incomplete partition type II
- SCC
- semicircular canal
- SNHL
- sensorineural hearing loss
- VA
- vertebral artery
SNHL is a major cause of childhood disability worldwide, with an estimated prevalence of 1 in 2000 neonates and 6 in 1000 children by 18 years of age.1 Early diagnosis and treatment of SNHL in children is critical because it is well recognized that a delay in identification of hearing impairment can adversely affect speech and language development, academic achievement, and social and emotional development.2 The institution of universal neonate hearing screening in the United States has altered the management of childhood SNHL by substantially lowering the average age at diagnosis from 24–30 months before the introduction of screening programs to 2–3 months.3
Cross-sectional imaging has become an integral tool in the clinical evaluation of SNHL. Otolaryngologists now routinely order CT or MR imaging examinations of the inner ear in infants and young children to identify a potential etiology for hearing loss, to define the anatomy of the temporal bone and the central auditory pathway, to identify abnormalities that may predict hearing loss progression or prognosis, and to identify additional intracranial abnormalities that may require further work-up and/or intervention.4
The aim of this 2-part series is to review the clinical and imaging evaluation of children presenting with SNHL, with an emphasis on aspects that neuroradiologists should know. Here we describe our typical clinical approach to imaging of children with SNHL, discuss the primary imaging techniques used for evaluation of the inner ear, review the classification of congenital inner ear malformations, and discuss common nonsyndromic causes of congenital SNHL. In the second part of the series, we will discuss syndromic congenital and acquired causes of SNHL that typically demonstrate inner ear abnormalities on imaging.
Work-Up of Childhood Hearing Loss and Choice of Imaging Technique
In most of the United States, neonate hearing screening is mandated by law. After a neonate fails hearing screening however, the evaluation process can be extremely variable because there are presently no definitive universal recommendations from federal agencies or medical professional societies for the comprehensive work-up of a neonate who fails hearing screening.
At our institution, a complete medical evaluation and a natural sleep diagnostic ABR test are performed. If a natural sleep study is not possible, a sedated or operative ABR is performed. If hearing loss is confirmed, the work-up centers around establishing a definitive cause, identifying potentially life-threatening conditions, and identifying other sensory deficits. This includes a complete ophthalmologic examination to rule out visual loss, retinitis pigmentosa (seen in Usher syndrome), or other eye problems; a 12-lead electrocardiogram to rule out prolonged QT or Jervell and Lange-Nielsen syndrome; genetic testing for connexin-related hearing loss; CMV testing of Guthrie card samples; genetics consultation; and cross-sectional imaging of the temporal bones and the central nervous system.4
Controversy exists as to the ideal algorithm for imaging children with newly diagnosed SNHL. Historically, CT has been the study of choice, but MR imaging has become increasingly popular as concerns mount over the risks associated with ionizing radiation exposure, particularly in children.5 Trimble et al6 have suggested that dual-technique imaging with high-resolution temporal bone CT and MR imaging identifies a substantially larger number of abnormalities in children being evaluated for cochlear implantation than either technique alone; however, whether the additional diagnostic yield of this strategy justifies the added cost of routinely performing both studies or even alters outcomes remains an area of debate.
Both modalities have their relative strengths and weaknesses. MR imaging avoids ionizing radiation and provides superior soft-tissue contrast. Advances in MR imaging technology, including high-field-strength units, improved coil technology, and parallel imaging, also allow increasingly high-resolution images of the inner ear and brain.7–10 High-resolution temporal bone CT is better for assessing bone detail and can be performed at lower cost and in less time, resulting in less frequent need for sedation or anesthesia compared with MR imaging.6,10
At our institution, MR imaging is the preferred initial imaging test performed in children with newly diagnosed SNHL. Our pediatric temporal bone MR imaging protocol, which is performed in approximately 20 minutes, includes either high-resolution FSE T2-weighted images (TR/TE/NEX, 1000/136 ms/1; echo-train length, 21; flip angle, 180°; FOV, 140 mm; matrix size, 192) or CISS images (TR/TE/NEX, 5.42–12.25/2.42–5.9 ms/1–2; flip angle, 50°–80°; FOV, 120–180 mm; matrix size, 256) through the temporal bones, as well as standard spin-echo T1-weighted images, FSE T2-weighted images, fluid-attenuated inversion recovery, and diffusion-weighted images through the entire brain. The CISS sequence generates near-isotropic voxels measuring 0.5–0.7 mm in length, which is adequate to visualize the anatomy of the fluid-filled inner ear and the nerves within the IACs. Intravenous contrast is not routinely administered in children unless there is a clinical suspicion of neoplasm or an infectious or inflammatory cause of hearing loss.
CT is reserved for the following cases: 1) The SCC defects or cochleovestibular anomalies are identified, so that the course of the facial nerve and cochlear anatomy can be delineated before cochlear implantation; 2) inner ear obstruction is evident, to determine whether the process is fibrous or osseous; 3) the IAC is narrow, precluding adequate visualization of the cochlear nerve, and it is necessary to determine patency of the bony cochlear nerve canal; or 4) if there is a conductive component to the hearing loss, to determine the integrity of the ossicular chain.4,11 Our routine pediatric temporal bone CT protocol includes both direct axial and coronal contiguous sequential acquisitions by using a collimation of 0.6–0.75 mm, 120 kV(peak), and variable mAs with current modulation (CARE Dose; Siemens Medical Systems, Erlangen, Germany) at a reference milliampere-second of 200. Scans extend from the top of the petrous apex to the mastoid tip in the axial direction and from the anterior tip of the petrous apex to the posterior margin of the mastoid in the coronal direction. In patients unable to tolerate direct coronal scanning, an axial spiral acquisition is performed, with axial and coronal image sets reconstructed at a section thickness of 0.6–0.75 mm.
Classification of Inner Ear Malformations
Inner ear malformations causing hearing loss are often discovered in infancy; however, some malformations, such as vestibular aqueduct enlargement, may not come to light until later in life. Malformations can range in severity from relatively mild dysplasias to complete inner ear aplasia. It is theorized that the spectrum of anomalies reflects interruptions to inner ear development occurring at different junctures during embryogenesis.12 Failure of otic placode formation during the third gestational week results in complete labyrinthine aplasia (Michel anomaly, Fig 1), whereas insults occurring later (during the seventh week) would only cause mild abnormalities (IP-II or Mondini dysplasia, Fig 2). In 2002, Sennaroglu and Saatci13 proposed a classification for cochleovestibular malformations that included, in order of decreasing severity: labyrinthine aplasia (Fig 1), cochlear aplasia (Fig 3), common cavity deformities (Fig 4), cystic cochleovestibular malformations (IP-I) (Fig 5), cochleovestibular hypoplasia (Fig 6), and IP-II (Fig 2), each of which is thought to result from an insult occurring at a progressively later stage of development. Features of each of these malformations are described in the Table.

Labyrinthine aplasia. Axial (A) and coronal (B) right temporal bone CT images demonstrate the complete absence of normal inner ear structures. Notice the diminished size of the inner ear edifice and absence of a well-formed internal auditory canal, which help to distinguish this from labyrinthitis ossificans.
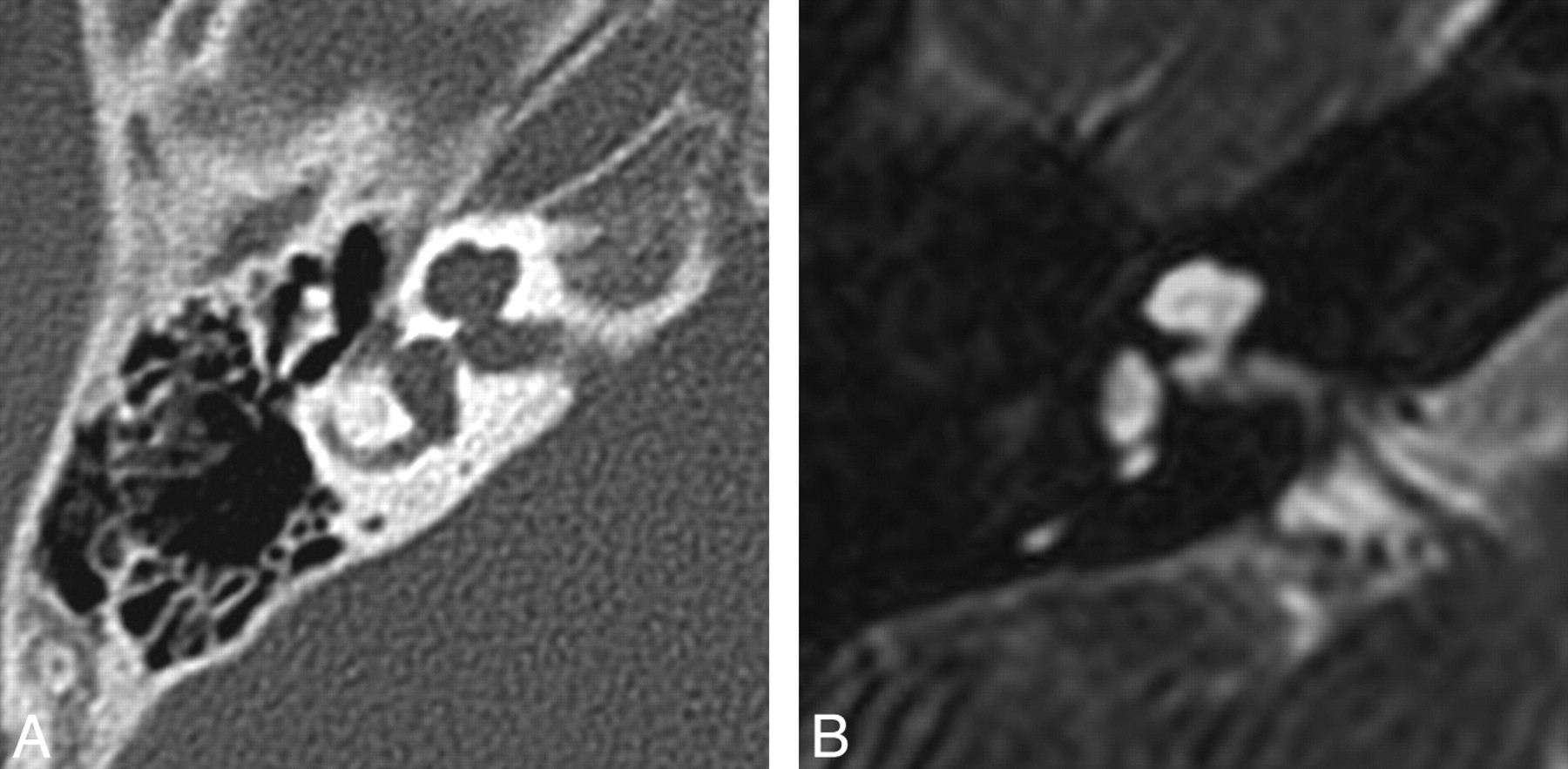
IP-II. Axial CT (A) and CISS (B) images through the right temporal bone demonstrate cochlear dysplasia with a deficient modiolus and fusion of the middle and apical turns, resulting in a bulbous-appearing cochlear apex. The vestibule and semicircular canals are normal, and the vestibular aqueduct is not enlarged.

Cochlear aplasia. Axial CT image through the right temporal bone demonstrates a dysplastic vestibule fused to an abnormal lateral SCC, with absence of the cochlea. Notice the small canal for the vestibular nerve (arrow).
Common cavity deformity. Axial CT (A) and CISS (B) images through the left inner ear demonstrate an enlarged cystic cavity representing the cochlea and vestibule without differentiation.
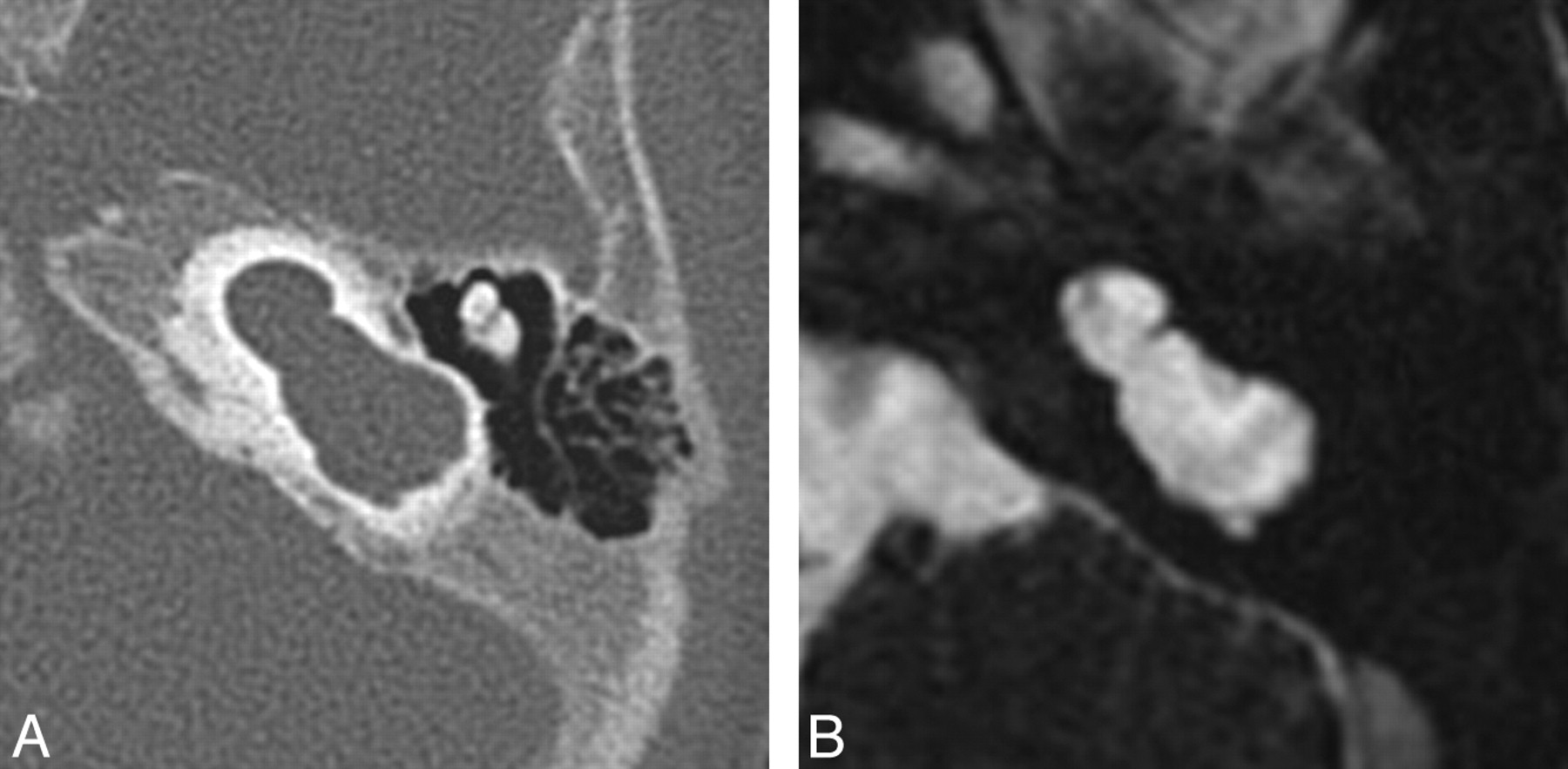
Cystic cochleovestibular anomaly (IP-I). Axial CT (A) and CISS (B) images through the right temporal bone demonstrate a cystic-appearing and featureless cochlea and vestibule. The cochlea and vestibule form a “figure 8” or “snowman” contour, with slightly increased separation compared with the common cavity deformity demonstrated in Fig 4.

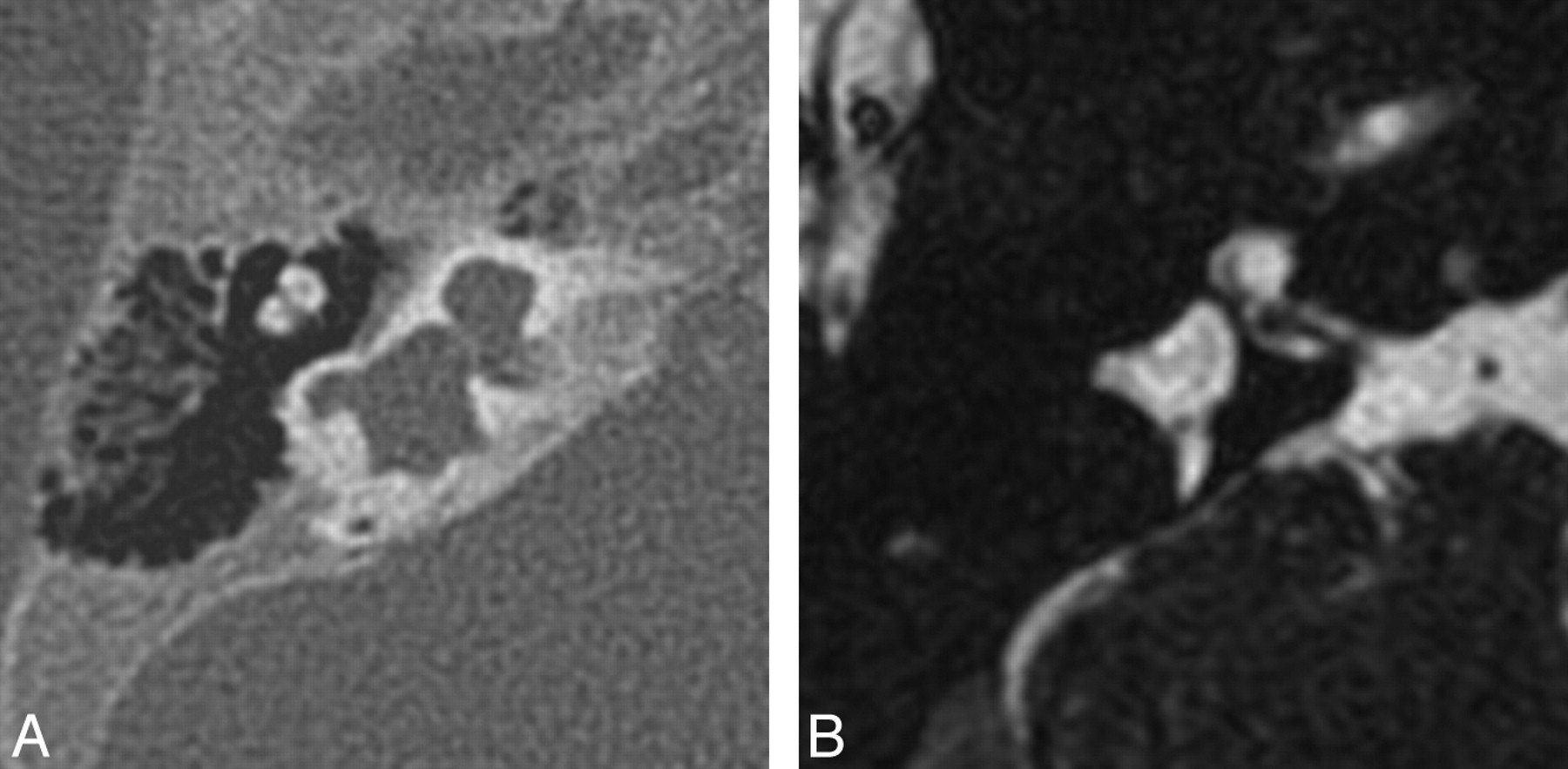
Cochlear hypoplasia. Axial CT (A) and FSE T2-weighted (B) images through the left temporal bone demonstrate a small cochlea, resembling a bud off of the IAC. The vestibule is also enlarged and dysplastic and is fused with a pocketlike lateral SCC.
Spectrum of congenital inner ear malformations as proposed by Sennaroglu and Saatci13
Vestibular aqueduct enlargement (also referred to as enlarged vestibular aqueduct syndrome) is the single most common inner ear anomaly seen in individuals with SNHL and is frequently seen in association with other inner ear anomalies, such as IP-II, vestibular enlargement, and SCC dysplasias. On CT, vestibular aqueduct enlargement is diagnosed when the aqueduct is >1.5 mm in width (roughly the diameter of the simultaneously visualized posterior SCC) at the midpoint between the common crus and its external aperture.14 MR imaging additionally demonstrates enlargement of the endolymphatic duct and sac (Fig 7 ), which may occasionally be seen without enlargement of the aqueduct.
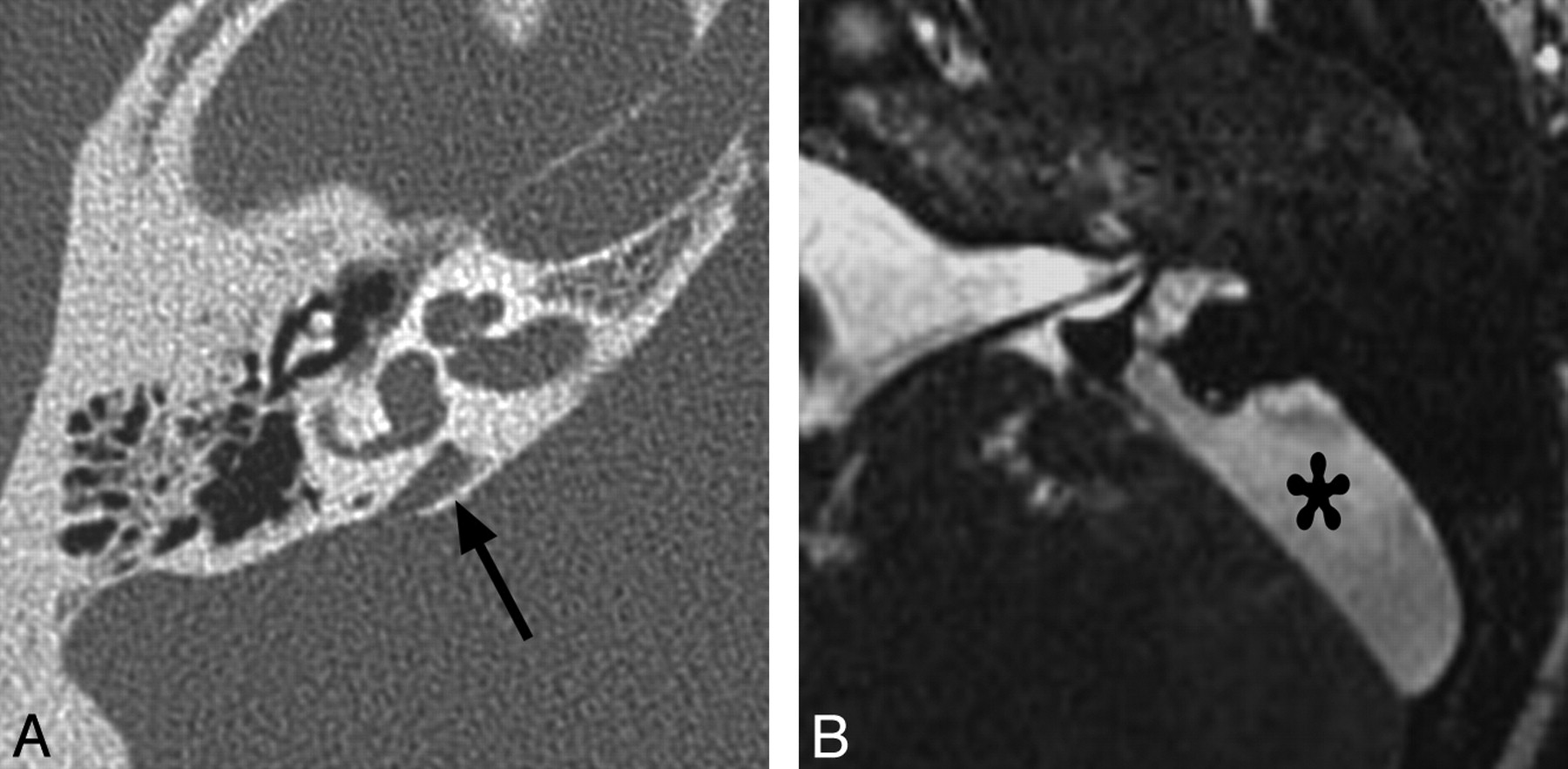
Enlarged vestibular aqueduct. A, Axial CT image through the right temporal bone demonstrates enlargement of the vestibular aqueduct, which has a fanlike configuration (arrow). Notice how the width of the aqueduct at its midpoint is larger than that of the adjacent SCCs. The cochlea is also mildly dysplastic (IP-II). B, Axial CISS image through the left temporal bone in a different patient demonstrates marked enlargement of the endolymphatic duct and sac (asterisk).
SCC malformations also frequently occur in association with other inner ear anomalies and follow a similarly predictable spectrum based on the timing of the causal growth disturbance. The SCCs begin as disk-shaped evaginations arising from the vestibular appendage in the sixth gestational week. The central portion of each disk is resorbed and replaced by mesenchyme, which results in formation of a characteristic semicircular duct. Failure of 1 of these disks to form results in the absence of the involved SCC, while incomplete absorption of the central portion of the disk results in a dysplastic or pocket-shaped SCC. The superior SCC is the first to form, followed by the posterior and then the lateral SCC. Therefore, superior and posterior SCC anomalies are almost invariably associated with anomalies of the lateral SCC, whereas abnormalities of the lateral SCC can occur in isolation (Fig 8 ).12 The 2 main exceptions to this rule are Waardenburg syndrome and Alagille syndrome, both of which may show absence of the posterior SCC without involvement of other SCCs.15,16 On axial CT images, subtle SCC abnormalities may be indicated by a small or enlarged lateral SCC bony island (Fig 9). Normally, the transverse diameter of this bony island measures between 2.6 and 4.8 mm.17
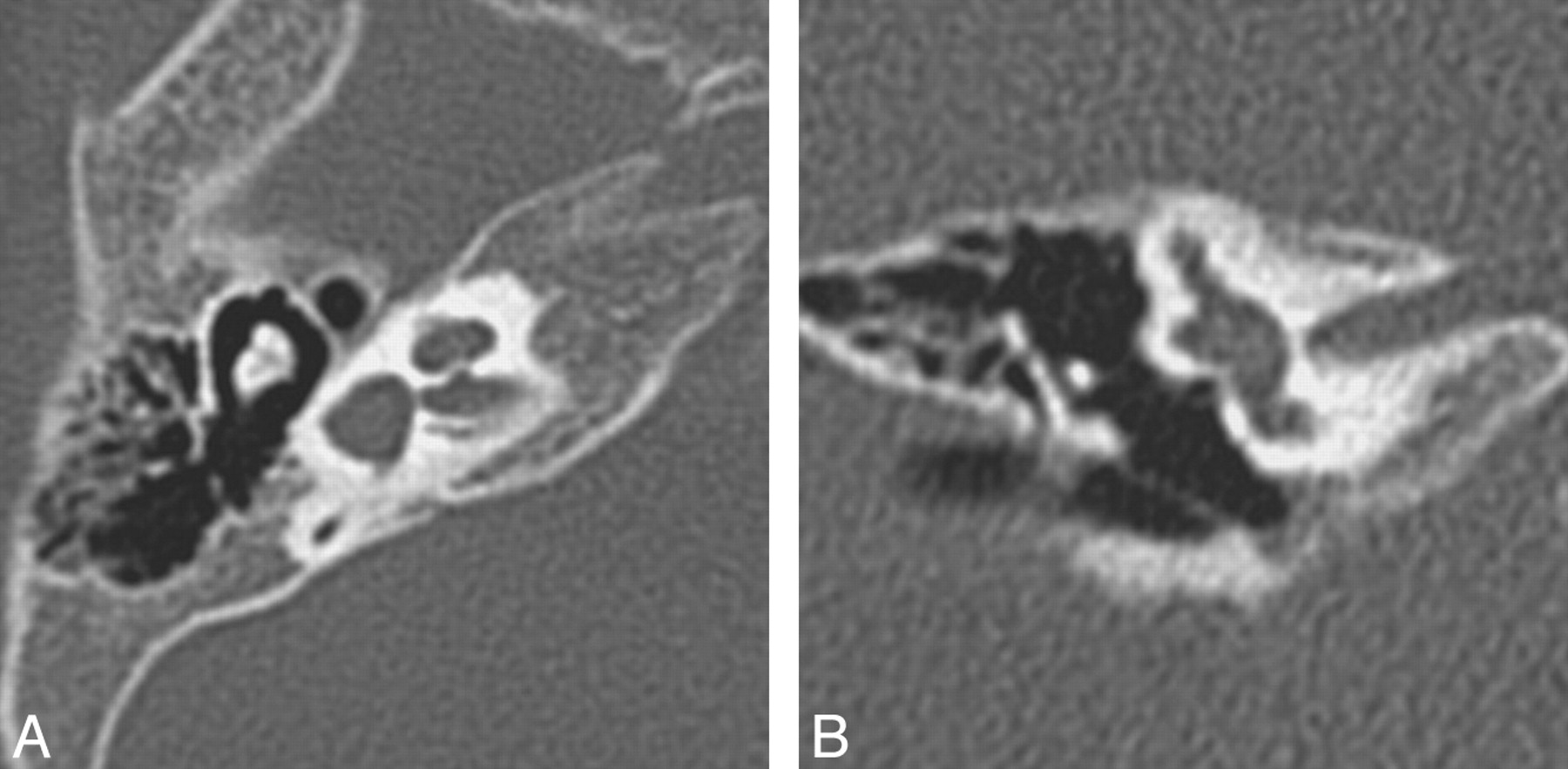
Lateral SCC dysplasia. Axial (A) and coronal (B) right temporal bone images demonstrate a dysplastic pocketlike lateral SCC, which is fused to the vestibule and missing its central bony island. On the coronal image, the lateral SCC is truncated. Both the superior and posterior SCCs are present.
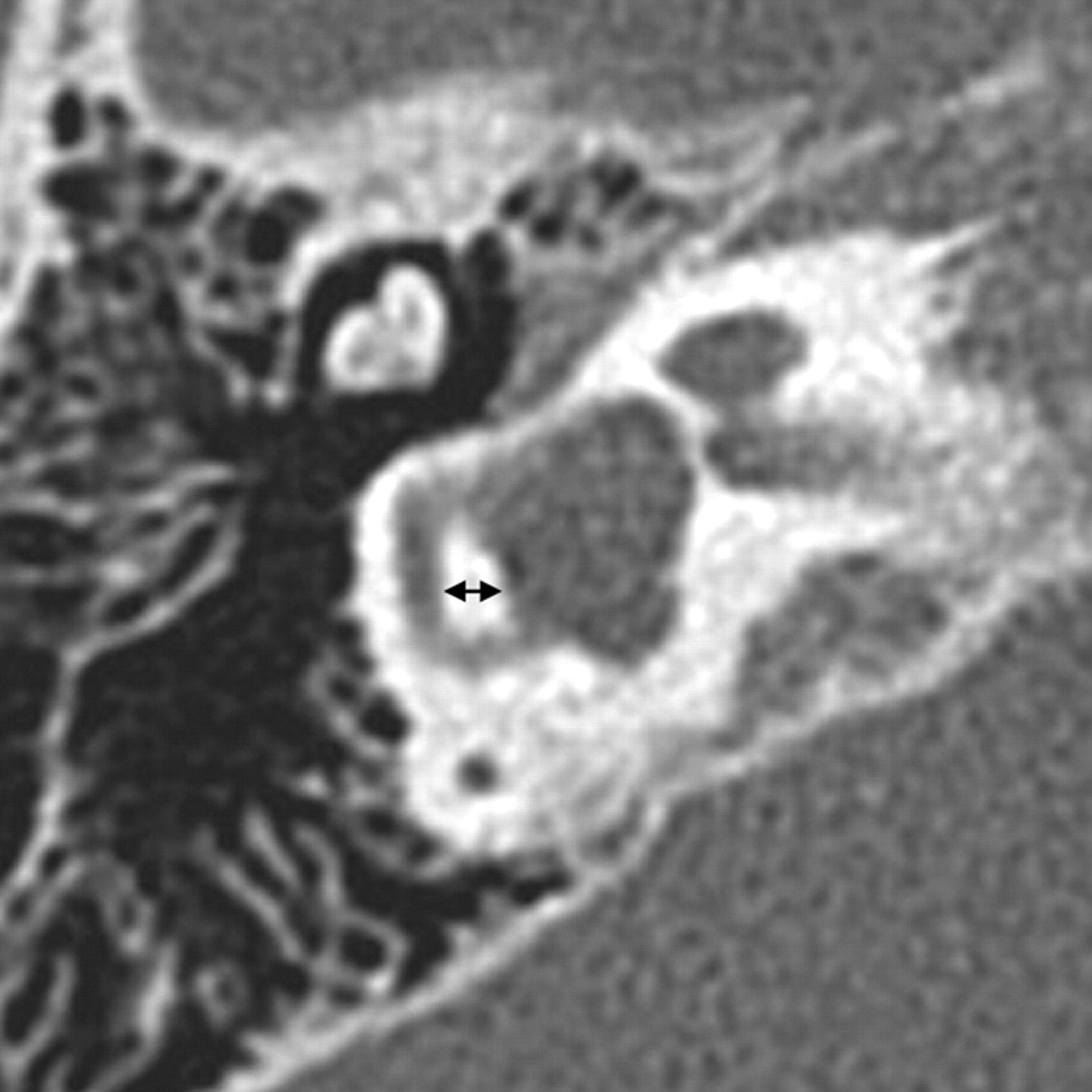
SCC dysplasia with a small lateral SCC bony island. The transverse diameter of the lateral SCC (double arrow) only measures 1.9 mm (normal, 2.6–4.8 mm). The vestibule is also enlarged and dysplastic, and the cochlea is hypoplastic.
Children with milder inner ear malformations (eg, vestibular aqueduct enlargement, IP-II, and partial SCC dysplasias) tend to perform better on tests of speech perception following cochlear implantation than children with other malformations (eg, common cavity or cochlear hypoplasia) or syndromes such as CHARGE syndrome,18,19 but even the presence of a severe malformation (provided a cochlea is present) does not preclude implantation. Identification of these anomalies is important in surgical planning, however, because the presence of cochlear malformations may make implant placement more challenging and also increases the risk for perilymph/CSF leak, postimplantation meningitis, and potential electrode misplacement, including placement into the IAC. In addition, the facial nerve course is aberrant in approximately 15%–32% of patients with cochleovestibular anomalies, a finding that influences surgical planning and occasionally precludes placement of the cochleostomy in the optimal position in relation to the round window.18,19
CND
CND refers to the absence or reduction in caliber of the cochlear nerve and is observed in 12%–18% of ears affected with SNHL.10,20 In pediatric patients, CND is usually congenital, but it can occasionally develop subsequent to birth due to atrophy of the nerve in patients who previously demonstrated normal hearing in the affected ear.21 Because cochlear implants are generally contraindicated in CND,22,23 it is important to identify this condition in children being considered for implantation.
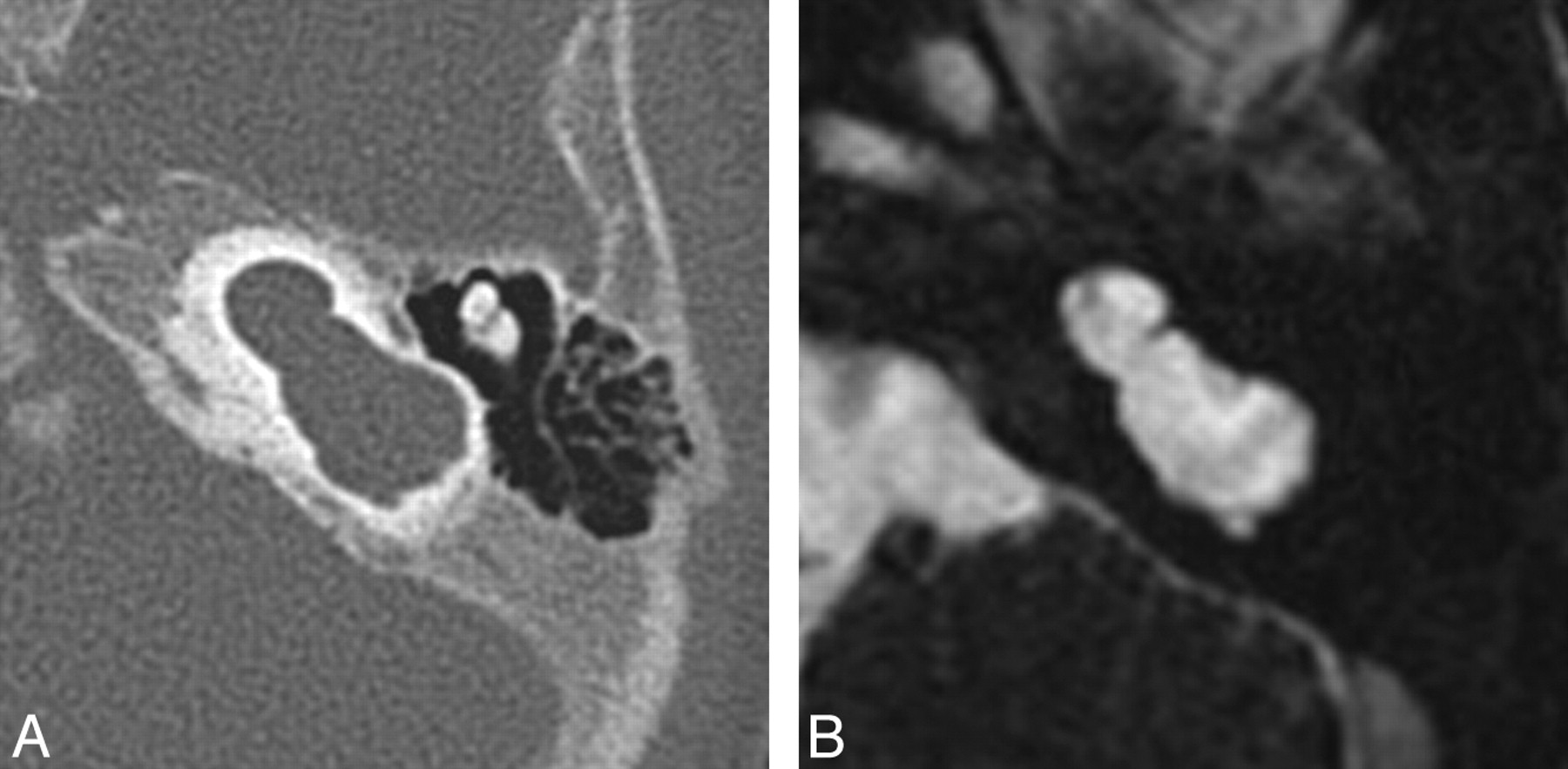
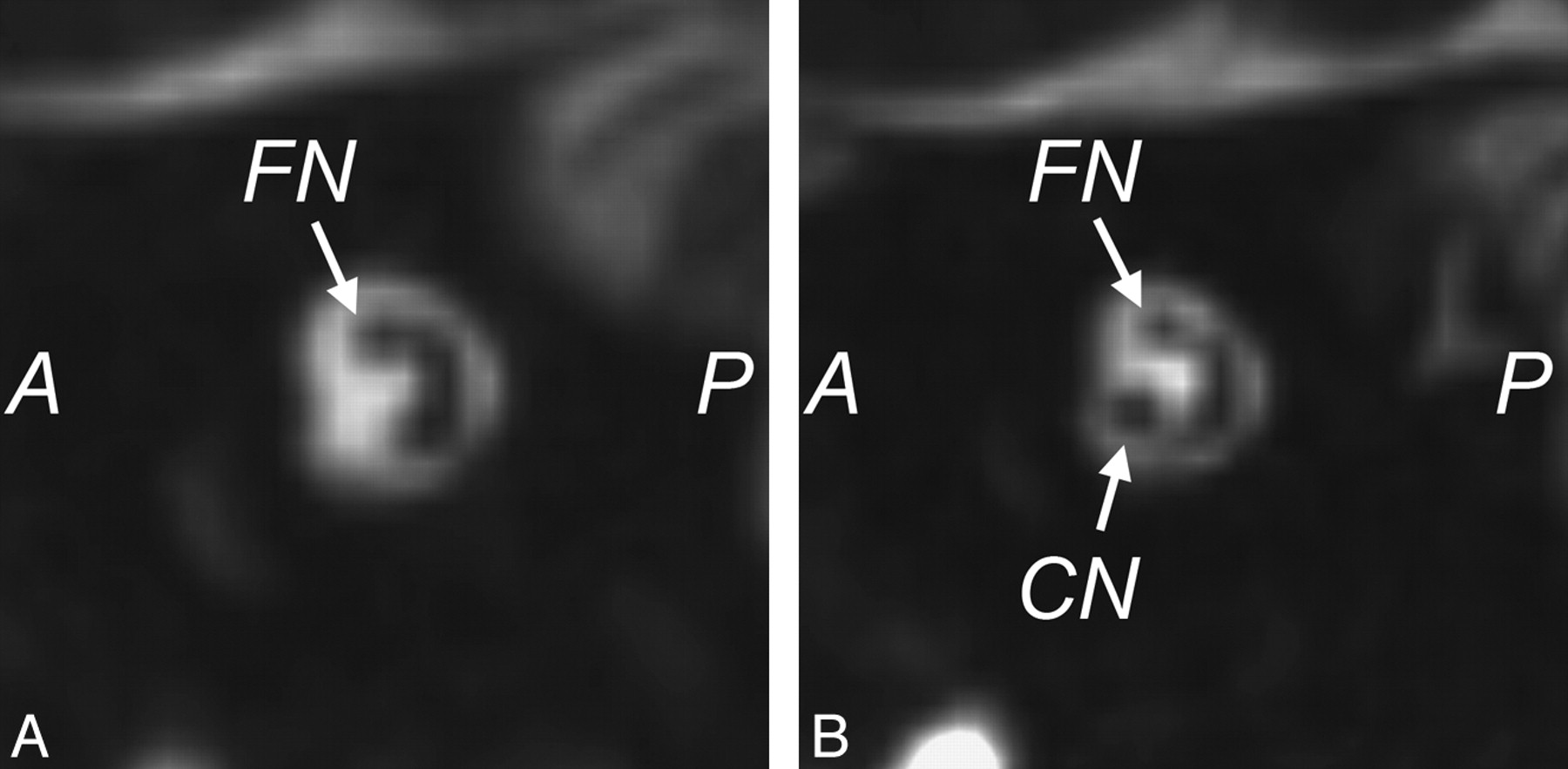
MR imaging is the most sensitive technique for diagnosing CND. On CISS images, the normal cochlear nerve is easily identifiable within the IAC. Oblique sagittal reconstructions oriented perpendicular to the long axis of the IAC are useful for visualizing the cochlear nerve, which is situated in the anterior inferior quadrant of the canal, as well as the intracanalicular segment of the facial nerve and the superior and inferior divisions of the vestibular nerve (Fig 10). CND can be seen in isolation (with the vestibular nerve divisions present) or in conjunction with aplasia of the vestibular nerve (complete absence of the eighth cranial nerve). Commonly, there will be associated narrowing of the IAC (defined as an IAC diameter of <4 mm), and it has been theorized that this occurs because the IAC depends on the presence of vestibulocochlear nerve cells to form normally.21,24

Cochlear nerve deficiency. A, Reconstructed sagittal CISS image through the right IAC demonstrates the absence of the right cochlear nerve. B, Compare with the sagittal image through the normal left IAC.
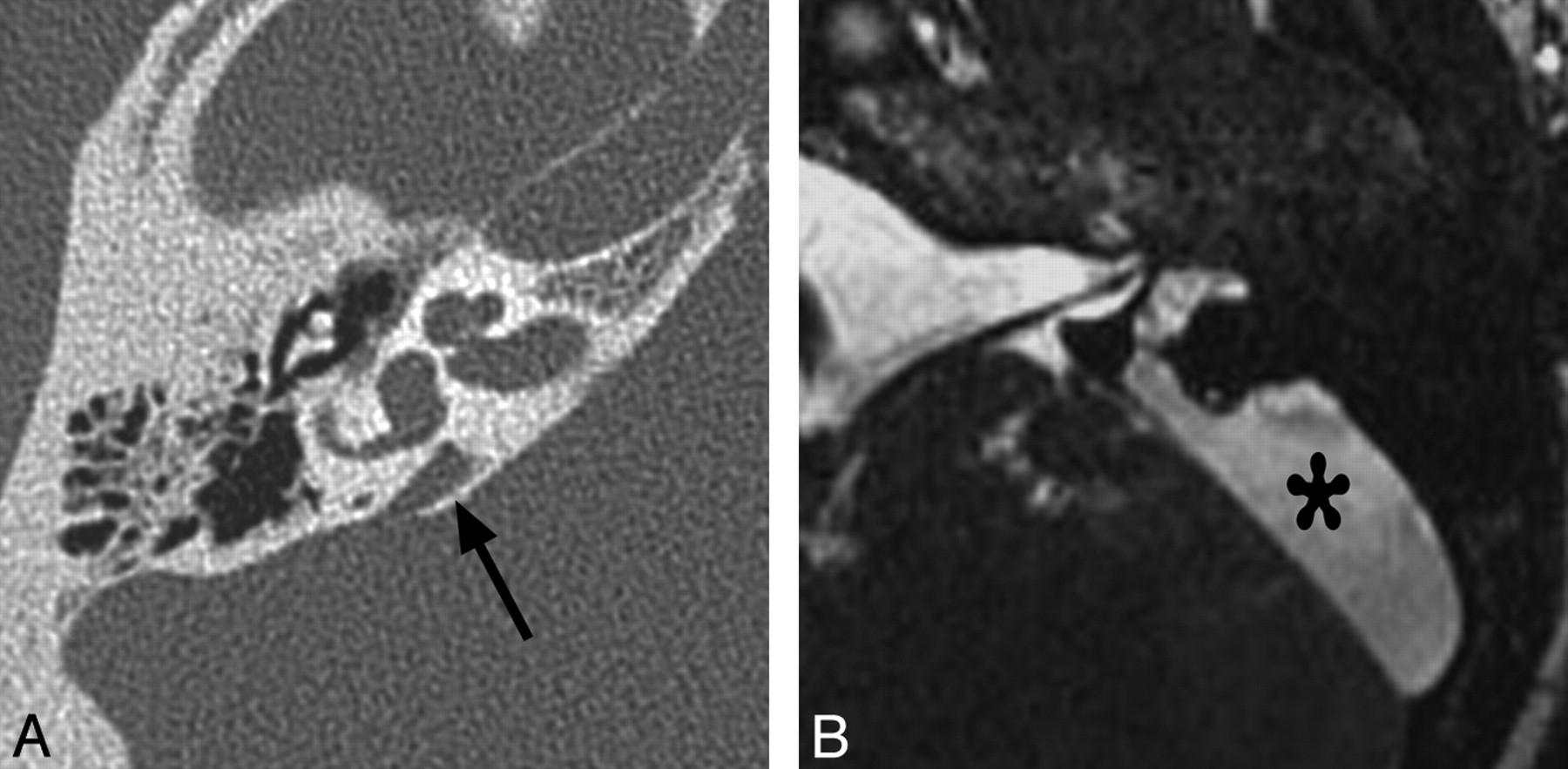
CT is less sensitive than MR imaging for the detection of CND but may demonstrate secondary signs, such as an absent or stenotic BCNC (normal between 1.4 and 3.0 mm)25 or a stenotic IAC (Fig 11 ). Children with narrow IACs on CT perform worse following implantation than those with normal caliber IACs, presumably because the cochlear nerve is likely to be absent or small when the IAC is narrow.19 Cochlear nerve−deficient ears may demonstrate normal caliber BCNCs in ≤23% of cases9 and normal sized IACs in ≤73% of cases.26

Bony cochlear nerve canal atresia in a patient with CND. A, Axial CT image through the right temporal bone of the same patient shown in Fig 10 demonstrates an atretic canal for the cochlear nerve at the base of the cochlea. B, Compare A with the normal-caliber BCNC on the left.
Congenital Hearing Loss
Congenital hearing loss is defined as hearing loss present at birth and is generally divided into genetic and nongenetic forms. It is estimated that in >50% of patients, SNHL can be linked to a genetic cause, of which approximately 75%–80% demonstrate autosomal recessive inheritance; 15%–20%, autosomal dominant inheritance; and 1%–2%, X-linked inheritance. Approximately 30% of inherited forms of hearing loss are syndromic, and the remaining 70% are considered nonsyndromic.27 To date, >300 syndromic forms of hearing loss have been described.28 Of the nongenetic forms of SNHL, an environmental cause can be identified in half and the remainder are considered idiopathic.29
Most children with congenital SNHL show normal inner ear morphology on CT and MR imaging, with congenital inner ear anomalies reported in only 20%–30%.10,20,30 This is because hearing loss is frequently due to isolated abnormalities on a cellular or microscopic level, which are not resolvable by current imaging techniques and which do not overtly affect the appearance of the bony otic capsule or membranous inner ear.
Below we discuss some nonsyndromic causes of congenital hearing loss, including both nonhereditary and hereditary etiologies. Syndromic hereditary forms of SNHL will be discussed in Part 2 of the series.
Nonhereditary Causes of Sensorineural Hearing Loss
Approximately half of the cases of congenital SNHL have a nonhereditary basis, and in roughly half of these, an underlying environmental cause can be identified. Etiologies include pre- and perinatal infections (CMV, rubella, measles, syphilis, and so forth), exposure to alcohol or ototoxic drugs (aminoglycosides, antineoplastic agents, and so forth), prematurity, hypoxic-ischemic injury, and hyperbilirubinemia.31 In general, these entities are not associated with gross inner ear abnormalities detectable by imaging.
CMV infection is the most prevalent environmental cause of prelingual hearing loss in the United States and has been implicated in approximately 10% of infants with congenital hearing loss and 34% of children with moderate-to-severe late-onset idiopathic hearing loss.32 Roughly 90% of neonates with congenital CMV have subclinical infections, but of these, approximately 10% develop significant permanent hearing loss. Furthermore, among children with clinically apparent congenital CMV infection, the prevalence of SNHL is roughly 30%.33 Hearing loss associated with congenital CMV can be unilateral, fluctuating, or progressive, and onset can be delayed for months or years. Inner ear abnormalities in deaf children with congenital CMV infection are not typical; however, associated brain abnormalities, which may include variable degrees of hemispheric white matter hyperintensity on T2-weighted images and cortical malformations, are present in ≤80%.34
Nonsyndromic Hereditary Causes of Hearing Loss
As already mentioned, most cases of hereditary SNHL are nonsyndromic. To date, >110 chromosomal loci and at least 65 genes causing nonsyndromic hearing loss have been identified.28 Of note, ≤50% of all cases of prelingual SNHL are attributable to mutations of the GJB2 gene, which encodes for the gap junction protein connexin 26.35 Most reports suggest that inner ear malformations are uncommon in children with GJB2 mutations, and several authors have suggested that routine imaging is not necessary in individuals with hearing GJB2-related deafness.36–38 This topic is controversial, however, because 1 report has suggested that 72% of subjects with biallelic GJB2 mutations have at least 1 temporal bone anomaly by CT imaging, with mild endolymphatic fossa enlargement and modiolar hypoplasia being the most common findings.39
By far, the most common inner ear malformation seen in children with nonsyndromic hearing loss is vestibular aqueduct enlargement. The prevalence of enlarged vestibular aqueducts in children with SNHL is estimated to be 10%–15%.40 The malformation is bilateral in approximately 90% of cases, and patients may present with profound congenital SNHL, progressive SNHL, or fluctuating SNHL. Initially, hearing loss may be of the high-frequency variety, and some patients may experience acute hearing decline related to episodes of minor head injury, overexertion, or barometric pressure changes (eg, related to air travel, deep sea diving, or Valsalva maneuver).14,41 In approximately 63% of nonsyndromic individuals with vestibular aqueduct enlargement, a mutation at the PDS locus on chromosome 7q31—which is the same gene responsible for Pendred syndrome—can be identified, suggesting a genetic basis for most cases of the malformation.41
Conclusions
Causes of hearing loss in the pediatric population are numerous and varied, often making evaluation and management of the condition quite challenging for otolaryngologists. Imaging plays a central role in the work-up of childhood SNHL because it depicts anomalies in the structures of the inner ear, frequently provides clues as to the etiology of the hearing loss, and may detect associated comorbid conditions. Although CT and MR imaging are both viable imaging choices, we prefer MR imaging due to its ability to directly assess not only the inner ear but also the cranial nerves and brain. In cases of congenital hearing impairment, inner ear malformations and CND are fairly common, and identification of these anomalies is critical when surgical intervention is being considered because they may complicate or even preclude cochlear implantation in a child with SNHL.
Indicates open access to non-subscribers at www.ajnr.org
References
- © 2012 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}