
Abstract
BACKGROUND AND PURPOSE: Acute hyperammonemic encephalopathy has significant morbidity and mortality unless promptly treated. We describe the MR imaging findings of acute hyperammonemic encephalopathy, which are not well-recognized in adult patients.
MATERIALS AND METHODS: We retrospectively reviewed the clinical and imaging data and outcome of consecutive patients with documented hyperammonemic encephalopathy seen at our institution. All patients underwent cranial MR imaging at 1.5T.
RESULTS: Four patients (2 women; mean age, 42 ± 13 years; range, 24–55 years) were included. Causes included acute fulminant hepatic failure, and sepsis with a background of chronic hepatic failure and post–heart-lung transplantation with various systemic complications. Plasma ammonia levels ranged from 55 to 168 μmol/L. Bilateral symmetric signal-intensity abnormalities, often with associated restricted diffusion involving the insular cortex and cingulate gyrus, were seen in all cases, with additional cortical involvement commonly seen elsewhere but much more variable and asymmetric. Involvement of the subcortical white matter was seen in 1 patient only. Another patient showed involvement of the basal ganglia, thalami, and midbrain. Two patients died (1 with fulminant cerebral edema), and 2 patients survived (1 neurologically intact and the other with significant intellectual impairment).
CONCLUSIONS: The striking common imaging finding was symmetric involvement of the cingulate gyrus and insular cortex in all patients, with more variable and asymmetric additional cortical involvement. These specific imaging features should alert the radiologist to the possibility of acute hyperammonemic encephalopathy.
Abbreviations
- ALT
- alanine transaminase
- AP
- alkaline phosphatase
- AST
- aspartate transaminase
- DWI
- diffusion-weighted imaging
- EEG
- electroencephalography
- FLAIR
- fluid-attenuated inversion recovery
- GCS
- Glasgow Coma Scale
- ICU
- intensive care unit
- IV
- intravenous
- N/A
- not applicable
- PT
- prothrombin time
- TPN
- total parenteral nutrition
Patients with acute hyperammonemic encephalopathy present with progressive drowsiness, seizures, and coma due to primary toxic effects of ammonia on the brain parenchyma.1 Prolonged hyperammonemia can lead to significant brain injury and long-term sequelae, such as intellectual impairment.1,2 Prompt recognition and treatment of hyperammonemia is, therefore, essential to avoid complications such as cerebral edema and brain herniation, which can prove fatal.2 In the pediatric population, acute hyperammonemic encephalopathy and its imaging findings have been well described as a result of inborn errors of metabolism (eg, urea cycle disorders or organic acidemias).3,4 In adults, this condition is more commonly encountered in very ill patients being treated in the ICU. Acute hepatic dysfunction is most frequently implicated, but other etiologies include portosystemic shunt surgery, drugs (eg, sodium valproate, asparaginase therapy, or chemotherapy), infections, hypothyroidism, multiple myeloma, and post-lung or bone marrow transplantation.1,2,5,–9
The radiologic findings of acute hyperammonemic encephalopathy are less well recognized in the adult literature, yet imaging may yield clues to the underlying diagnosis. The purpose of this study was to describe the MR imaging features of adult hyperammonemic encephalopathy.
Materials and Methods
The institutional review board of our hospital approved this retrospective study and waived informed consent. Our study included consecutive patients treated between 2005 and 2009, identified from a prospectively collected data base, with documented hyperammonemic encephalopathy. Clinical records, biochemical laboratory indices, and EEG reports were reviewed for each patient.
All patients underwent cranial MR imaging at 1.5T (Signa HDXT, GE Healthcare, Milwaukee, Wisconsin; or Magnetom Avanto, Siemens Medical Systems, Iselin, New Jersey). Routine spin-echo sagittal T1-weighted, T2-weighted, FLAIR, ultrafast spoiled gradient-echo, and DWI sequences were performed in all 4 cases. Routine postcontrast T1-weighted imaging in the axial and coronal planes was performed in 2 patients after IV injection of 0.1 mmol/kg of gadobutrol (Gadovist; Bayer Schering Pharma, Berlin, Germany). The MR imaging features were analyzed by 1 neuroradiologist (J.M.U.-K.-I), who recorded the extent of signal-intensity abnormalities on FLAIR, T2-weighted sequences, and DWI. The pattern of involvement, including cortical and subcortical white matter; distribution; symmetry; enhancement; and involvement of other sites, such as the basal ganglia, thalami, and posterior fossa structures, were recorded.
Results
The salient demographics, clinical data, and MR imaging features of the 4 patients with hyperammonemic encephalopathy are summarized in the On-line Table.
Patients
Four patients (2 women; mean, age 42 ± 13 years; range, 24–55 years), all in the ICU, were included in this study. All patients presented with seizures and reduced level of consciousness (GCS scores ranging from 3 to 8). Additionally, 1 patient had abnormal posturing. Plasma ammonium levels ranged from 55 to 168 μmol/L (normal range, 0–34 μmol/L). EEG was performed in 3 cases on days 2–4 postictus and showed nonspecific generalized encephalopathic disturbance of cerebral activity in both hemispheres (On-line Table).
Patient 1 was post–heart-lung transplantation for severe pulmonary arterial dilation, pulmonary insufficiency, and mitral valve prolapse (Figs 1 and 2). Her postoperative course in the ICU was very protracted, complicated by cardiac failure, pulmonary edema, chest infection, sternal wound sepsis, and venous thrombosis. She was on a standard immunosuppressive regimen (methylprednisolone, azathioprine, and cyclosporine and mycophenolate mofetil) and had been on total parenteral nutrition.

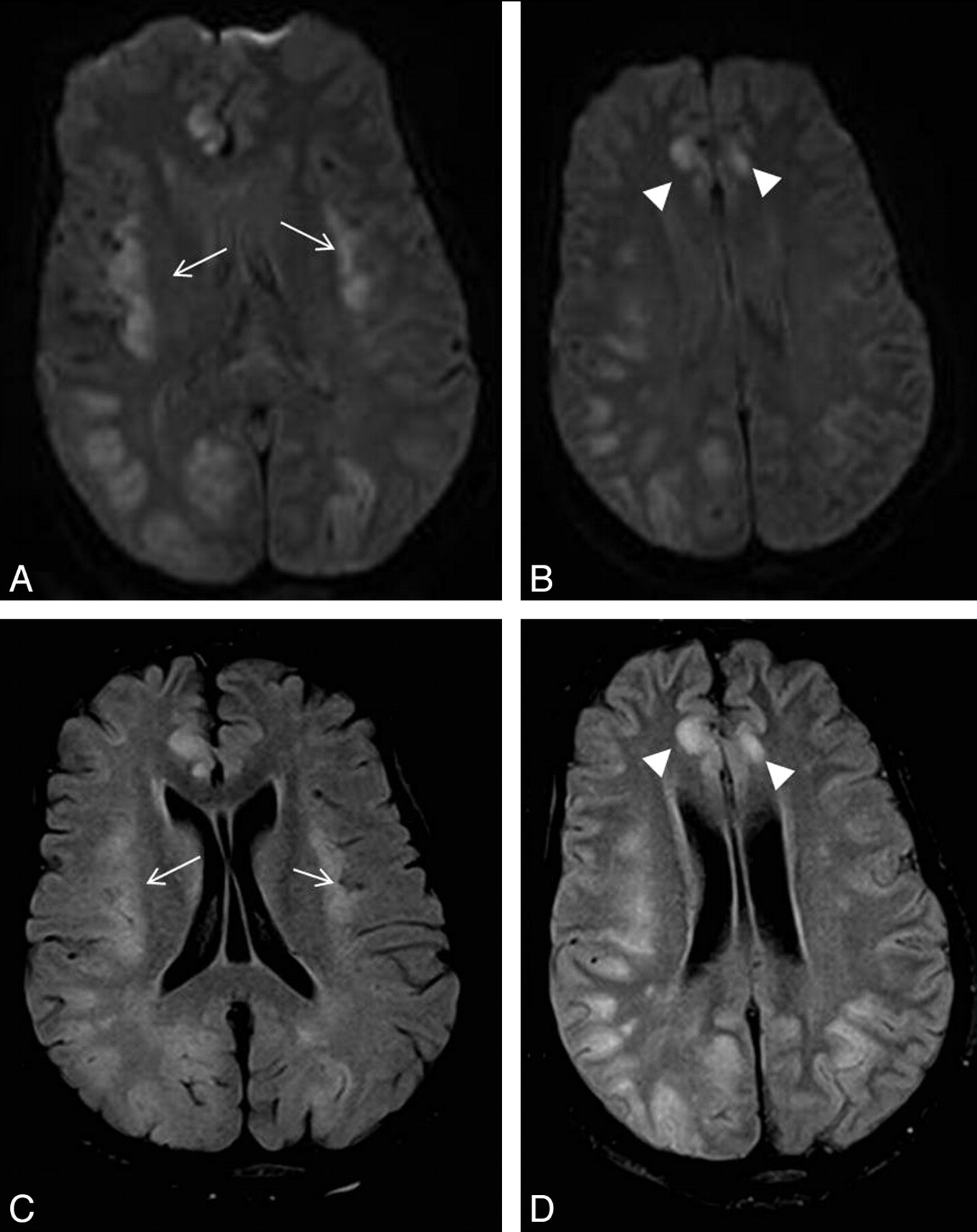
Patient 1. A 24-year-old woman with complicated (cardiac failure, pulmonary edema, chest infection, sternal wound sepsis, and venous thrombosis) and very protracted postoperative course in the ICU post–heart-lung transplantation. She was on a standard immunosuppressive regimen and had been on total parenteral nutrition. Five weeks after surgery, she deteriorated neurologically with seizures and decreased level of consciousness. Her plasma ammonium level was 168 μmol/L. DWI (A and B) and FLAIR images (C and D) show symmetric abnormal signal intensity in the insular (arrows) and cingulate cortices (arrowheads) bilaterally. Extensive signal-intensity change involving both the cortex and subcortical white matter is also seen in right temporo-occipital and left occipital lobes.

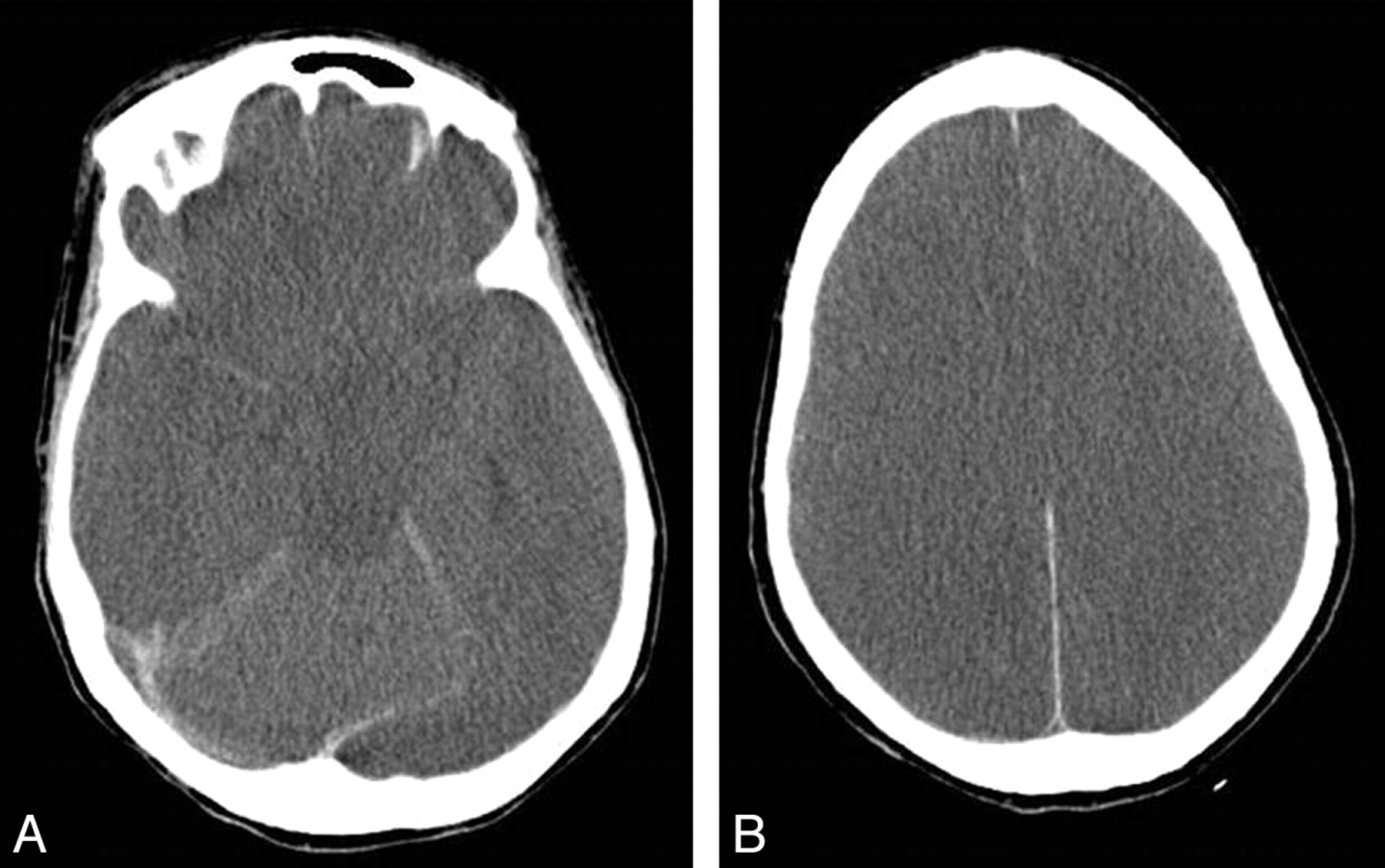
Patient 1. Follow-up brain CT scan after 7 days in patient 1, who continued to deteriorate neurologically despite therapeutic measures. Repeat plasma ammonium level decreased to 110 μmol/L. A, Effacement of suprasellar cisterns in keeping with bilateral uncal herniation is seen. B, Image at a level above the lateral ventricles shows widespread loss of cortical sulci and gray-white differentiation, in keeping with cerebral edema. The patient died soon after the study was performed.
Patient 2 presented with fulminant acute hepatic failure following an overdose of acetaminophen with a background of chronic alcohol abuse (Fig 3). Liver function test findings were grossly abnormal (bilirubin, 81 μmol/L; ALT, 3492 IU/L; AP, 178 IU/L; AST, 20,872 IU/L; PT, 83 seconds).

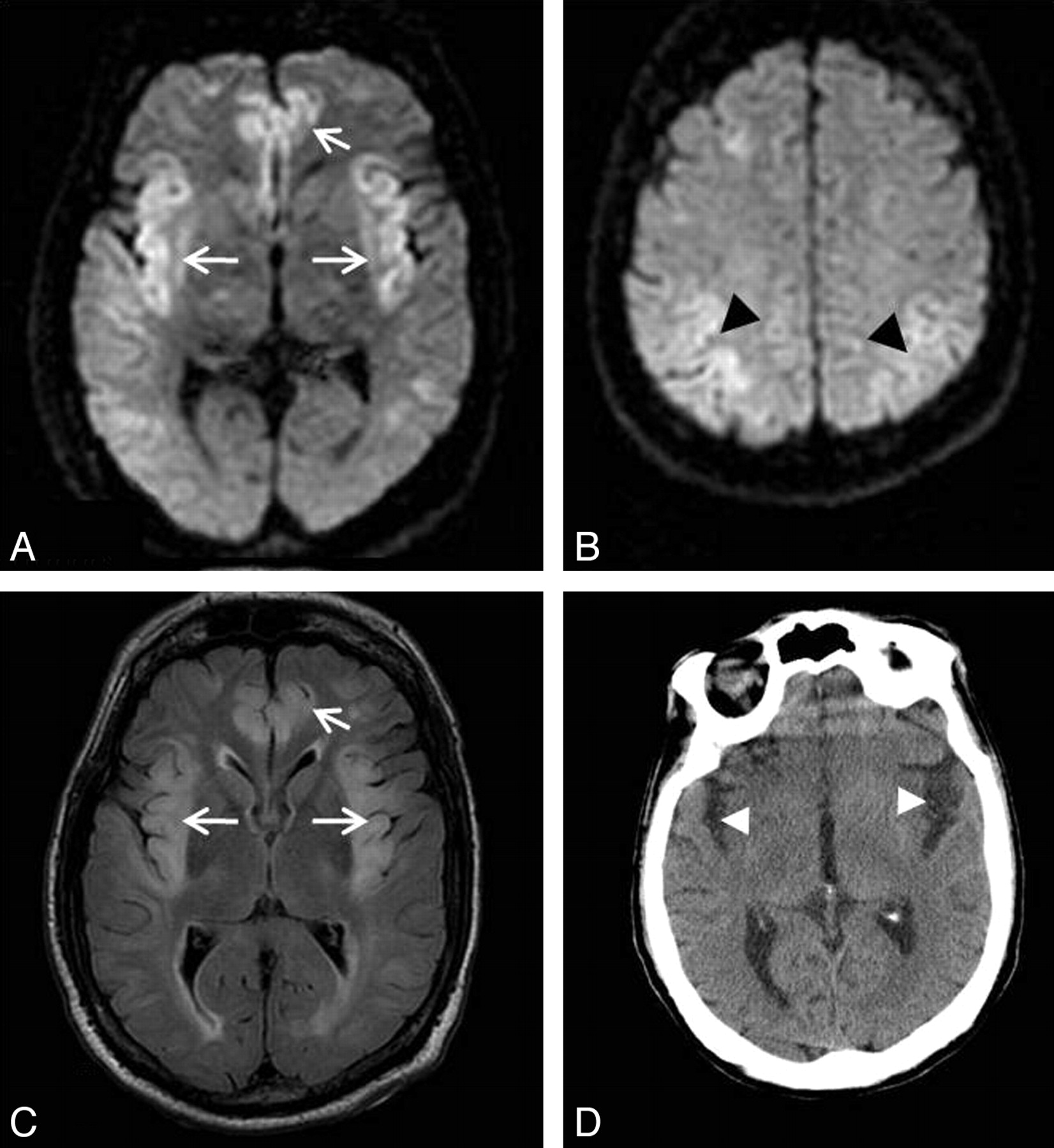
Patient 2. A 48-year-old man with fulminant acute hepatic failure due to acetaminophen overdose with a background of chronic alcohol abuse, His ICU stay was complicated by sepsis due to pneumonia and Clostridium difficile diarrhea. On day 8 post−ICU admission, he deteriorated neurologically with a decreased level of consciousness, abnormal posturing, and seizures. DWI (A and B) and T2-weighted images (C and D) show symmetric abnormal high signal intensity in the insular and cingulate cortices (open arrows). High signal intensity is also seen involving the cortex of the temporoparietal lobes bilaterally, bilateral thalami (arrowheads), and brain stem (closed arrow). His plasma ammonium level was 102 μmol/L. The patient's condition deteriorated further, and he died the next day without repeat imaging.
Patient 3 presented with severe sepsis of unknown origin (blood cultures positive for Enterococcus species) with a background of schizophrenia, chronic hepatitis C, portal hypertension, and esophageal varices (Fig 4). Liver function test results were mildly deranged (bilirubin, 20 μmol/L; ALT, 63 IU/L; ALP, 201 IU/L; AST, 271 IU/L; PT, 50 seconds).

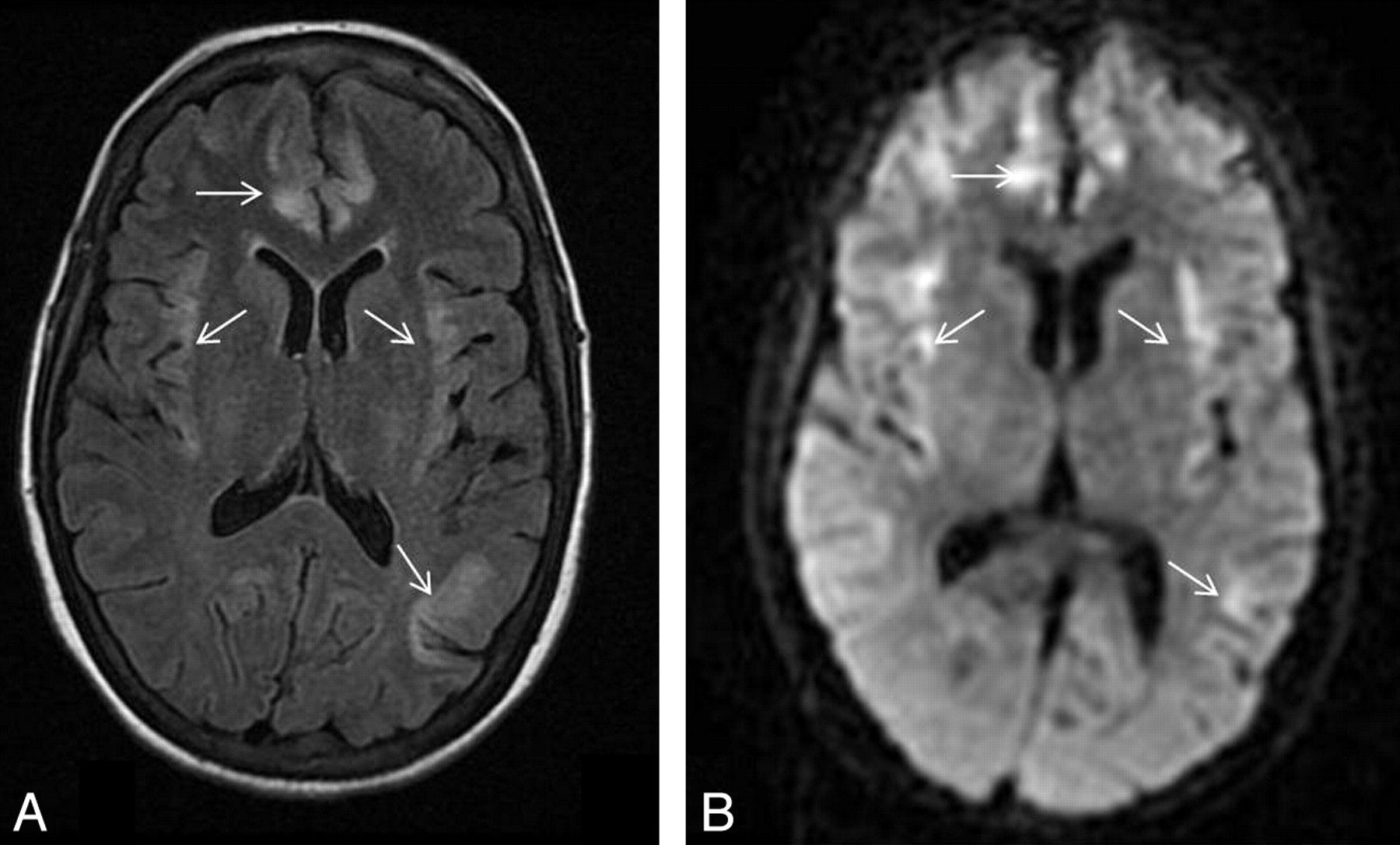
Patient 3. A 55-year-old man with severe sepsis of unknown origin (positive blood cultures for Enterococcus species) with a background of schizophrenia, chronic hepatitis C, portal hypertension, and esophageal varices. The patient had a reduced level of consciousness and seizures. His plasma ammonium level was 82 μmol/L. A−C, DWI (A and B) and FLAIR image (C) show high signal intensity in the insular and cingulate cortices (arrows) and the perirolandic cortex bilaterally (black arrowheads). There is also subtle high signal intensity in the subinsular regions. With conservative therapeutic measures, the patient gradually improved neurologically, his plasma ammonium level decreased to 50 μmol/L, but he had significant residual intellectual impairment. D, Follow-up brain CT after 1 month showed widening of Sylvian fissures bilaterally (white arrowheads) in keeping with insular atrophy.
Patient 4 was admitted with severe sepsis related to her tunneled central dialysis line (Fig 5). She had undergone liver transplantation 13 years prior for nonalcoholic steatohepatitis, chronic graft rejection, worsening liver function, and hepatorenal syndrome, for which she had been undergoing hemodialysis. Her immunosuppressant regimen included tacrolimus and cyclosporin. Liver function test results were mildly deranged (bilirubin, 68 μmol/L; ALT, 77 IU/L; ALP, 97 IU/L; AST, 414 IU/L; PT, 46 seconds).

Patient 4. A 42-year-old woman was admitted with sepsis related to her central dialysis line, with a background of liver transplant, chronic graft rejection, and hepatorenal syndrome. Her immunosuppressant regimen included tacrolimus and cyclosporin. Her condition deteriorated with respiratory failure, hypotension, seizures, and a drop in the GCS score. Her plasma ammonium level was 55 μmol/L. FLAIR image (A) and DWI (B) show abnormal signal intensity in the cortex involving the bilateral insular and cingulate and left posterior temporal lobes (arrows). The patient made an excellent recovery without significant neurologic deficit with supportive therapeutic measures.
Imaging Findings
MR imaging was performed on days 1–4 post-onset of symptoms. Bilateral symmetric involvement of the insular cortex and cingulate gyrus was present in all 4 patients on FLAIR and T2-weighted imaging sequences (Figs 1 and 3⇑–5). Additional involvement of parietal, frontal, temporal, or occipital cortices was seen in all patients but was much more variable in extent and was also asymmetric (On-line Table and Figs 1 and 3⇑–5). Most areas of signal-intensity abnormality also showed corresponding areas of restricted diffusion (Figs 1 and 3⇑–5). Intravenous contrast medium was administered in 2 cases, and no abnormal enhancement was seen in either case.
MR imaging performed in patient 1 also showed increased T2 and FLAIR signal intensity with restricted diffusion in the subcortical white matter in the temporal and occipital lobes, a pattern not seen in the 3 other cases (Fig 1). MR imaging in patient 2 only showed increased T2 and FLAIR signal intensity in the basal ganglia, thalami, and midbrain bilaterally without restricted diffusion (Fig 3). MR imaging in patient 3 showed involvement of the perirolandic cortex bilaterally (Fig 4).
In the clinical radiologic reports, the possibility of hyperammonemic encephalopathy was raised in 3 cases. The differential diagnosis given in the clinical radiologic reports included posterior reversible encephalopathy syndrome, seizure activity, metabolic and hepatic encephalopathy, and diffuse hypoxic-ischemic injury.
Treatment and Outcome
Treatment in the 4 patients included lactulose; dietary protein restriction; discontinuation of TPN, N-acetylcystine, propofol, and other supportive measures such as mannitol to reduce cerebral edema; antiseizure medications, such as phenytoin and phenobarbital; and broad-spectrum antibiotics and antifungal agents for superadded infection. Hemodialysis, a potential treatment used in severe cases, was not used in any of the 4 patients.1
Two patients died. The condition of patient 1, who had the highest plasma ammonia level at 168 μmol/L, deteriorated further clinically despite therapeutic measures and plasma ammonium levels decreasing to 110 μmol/L. Repeat CT imaging showed evidence of severe cerebral edema and brain herniation (Fig 2). The condition of patient 2 deteriorated further after admission to the ICU, and he died 2 days later without repeat imaging. Of the 2 survivors, patient 4 made an excellent recovery without significant intellectual or neurologic deficit. Patient 3 was discharged from the ICU 2 weeks later with a GCS score of 11 (E3, V2, M6) and significant intellectual impairment. Repeat CT imaging showed widening of the Sylvian fissures in keeping with atrophy (Fig 4).
Discussion
All our 4 patients were very ill and were being treated in the ICU. In all cases, extensive cortical signal-intensity changes, with associated restricted diffusion, were seen; these features are in keeping with early changes seen in hyperammonemic encephalopathy. These early imaging findings are not well-recognized in the adult literature; for example, in their comprehensive review of MR imaging findings in hepatic encephalopathy published in the American Journal of Neuroradiology in 2008, Rovira et al10 only discussed MR spectroscopic findings and late changes such as cerebral edema and brain herniation in the setting of fulminant hepatic failure and hyperammonemia.
Bilateral involvement of the insular cortex and cingulate gyrus was a strikingly common feature, seen in all our 4 patients, and these findings have also been described in several other cases, mainly in the pediatric literature.4,11
Bindu et al4 reported 3 cases of hyperammonemic encephalopathy in children due to various causes including infantile citrullinemia, acute hepatic encephalopathy, and proximal urea cycle disorder, with similar cortical abnormalities and involvement of the insular and cingulate cortices. Takanashi et al11 described 3 cases of acute hyperammonemic encephalopathy arising from late-onset ornithine transcarbylamase deficiency, all with evidence of injury to the cingulate and insular cortices, and they described sparing of perirolandic and occipital cortices.
Involvement of brain regions other than the insular or cingulate gyrus was clearly more variable. Unlike the cases described by Takanashi et al,11 involvement of the occipital or perirolandic cortex was seen in 2 of our cases (patients 1 and 3, respectively). Additional sites of involvement, such as the basal ganglia, brain stem, and thalami, were seen in 1 patient only (patient 2). Brain stem involvement was also reported by Arnold et al,12 who described extensive cortical changes in an adult patient with hepatic encephalopathy, which were initially reported as diffuse ischemic injury. Although the involvement of insular and cingulate cortices was not emphasized, this can clearly be seen on the MR images provided in their article.
Other causes of hyperammonemic encephalopathy with similar imaging findings include valproic acid−induced hyperammonemia and adult-onset citrullinemia.7,13,14
Patient 1 also had signal-intensity change involving the subcortical white matter in addition to the cortical abnormalities; this pattern is recognized in the pediatric literature in patients with inborn errors of metabolism, in which subcortical white matter involvement may often predominate over cortical signal-intensity changes.3 Thus, it is plausible that both the cortical and subcortical changes are due to the same underlying etiology. Subcortical white matter involvement was not seen in 2 of the other patients in our series, though subtle subinsular changes were seen in patient 3. It is possible that this pattern is only seen at higher plasma ammonium levels, as in the case of patient 1, who had the highest plasma ammonium level at 168 μmol/L.
Three of our cases were related to hepatic failure (1 acute fulminant hepatic failure and 2 others with sepsis and a background of chronic hepatic failure), and 1 case was seen post–heart-lung transplantation. Hyperammonemic encephalopathy post–orthotopic lung transplantation without evidence of hepatic dysfunction has been well-described; in the series by Lichtenstein et al,2 4 of 6 cases resulted in fulminant and fatal cerebral edema. The authors postulate a multifactorial etiology including immunosuppressive drugs, TPN, and medical stressors such as major gastrointestinal complications.
Most of the patients in our series had a number of systemic problems, and we are unable to completely exclude the fact that the MR imaging changes could potentially be partly due to other factors such as hypoxic injury or seizure activity. However, the changes in the cingulate and insular gyri were very symmetric, more in favor of a toxic encephalopathy. Moreover, it is very plausible from the striking similarity among our cases and others reported in the literature, that the imaging pattern described is directly related to hyperammonemia rather than to seizure or hypoxic injury or to the various causative underlying conditions, whether hepatic failure, post-lung transplantation, or inborn errors of metabolism.15
Ammonia is produced mainly in the gastrointestinal tract as a by-product of protein digestion and bacterial metabolism.16 It is metabolized primarily in the liver as urea through the urea cycle, where several key enzymes include carbamyl phosphate synthetase and ornithine transcarbamylase.1,16 When the metabolic capacity of the liver is overwhelmed, elimination becomes dependent on the kidneys, brain, and skeletal muscle. In the brain, astrocytes rapidly metabolize ammonia and glutamate to glutamine via the glutamine synthetase pathways; however, this is physiologically costly and the subsequent rapid increase in cellular osmolarity causes astrocyte swelling and loss.17 Inflammatory cascades, apoptosis, and various metabolic pathways are triggered, resulting in elevation in lactate, loss of cerebral autoregulation, and cerebral edema.16 This mechanism is supported by MR spectroscopy, which has shown high glutamine concentration in affected areas in patients with urea cycle disorders.18 It is not known why the insular and cingulate cortices seem particularly susceptible to the toxic effects of hyperammonemia. Most interesting, Takanashi et al19 described 3 cases of neonatal hyperammonemic encephalopathy from proximal urea cycle disorders with a slightly different pattern, with involvement of the lentiform nuclei, insular sulci, and perirolandic regions. They postulated that these may be more metabolically active brain areas in the neonatal period and suggested that hypoperfusion and ischemia may be playing a part in the mechanism of injury in the setting of raised intracranial pressure.
Cortical changes in hyperammonemic encephalopathy have been shown to be potentially reversible14 or to result in variable amounts of atrophy in the cingulate and insular cortex after treatment (seen in patient 3).4,11,13 Our findings support the general view that these cortical changes appear to be early imaging findings and potentially reversible if aggressive treatment is instituted.4,11 The extent of injury may be dependent on the severity and duration of the hyperammonemia and predisposing susceptibility to the metabolic insult.2,11 Our data also suggest that the plasma ammonium levels may reflect the extent of injury. While coma, cerebral edema, and brain herniation are usually seen at higher levels of plasma ammonia (at least 4 times the normal range), early clinical manifestations of hyperammonemia (eg, anorexia, irritability, lethargy, vomiting, somnolence, disorientation) can be seen with lower plasma ammonia levels of ≥60 μmol/L, and our findings suggest that early MR imaging changes can be seen at such levels.20 Patient 4, with the lowest plasma ammonium level (55 μmol/L), had the least extensive MR imaging changes and made an excellent recovery without significant neurologic deficit. Patient 3, with plasma ammonium levels of 82 μmol/L, survived with severe intellectual impairment. Both patients with plasma ammonium levels of 102 μmol/L (patient 2) and 186 μmol/L (patient 1) did not survive, and patient 1, with the highest plasma ammonium levels, proceeded to fulminant cerebral edema and brain herniation as demonstrated on CT.
Conclusions
We report a specific pattern of imaging findings, with extensive cortical injury, including bilateral symmetric involvement of the insular and cingulate cortices, which should alert the radiologist to the possibility of hyperammonemic encephalopathy. We postulate that with more severe injury, involvement of subcortical white matter, basal ganglia, thalami, and brain stem can also be seen. This pattern has been previously described in children with metabolic disorders but is less well-recognized in adults.
Footnotes

Indicates article with supplemental on-line table.

References
- Received June 25, 2010.
- Accepted after revision July 19, 2010.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Pearls & Oy-sters: Status Epilepticus and Cerebral Edema From Hyperammonemia Due to Disseminated Ureaplasma and Mycoplasma Species
- Hyperammonaemia syndrome in disseminated Ureaplasma parvum infection
- A 73-Year-Old Woman With Hyperammonemic Encephalopathy Caused by Noncirrhotic Congenital Portosystemic Shunts
- Claustrum hyperintensity: a rare radiological correlate in Niemann-Pick disease
- Non-cirrhotic hyperammonaemia: are we missing the diagnosis?
- Pearls & Oy-sters: Chemotherapy-associated hyperammonemic encephalopathy
- Multifactorial non-cirrhotic hyperammonaemic encephalopathy
- Multimodality MR Imaging Findings of Low-Grade Brain Edema in Hepatic Encephalopathy
- Reply: