
Abstract
SUMMARY: A variety of congenital syndromes affecting the face occur due to defects involving the first and second BAs. Radiographic evaluation of craniofacial deformities is necessary to define aberrant anatomy, plan surgical procedures, and evaluate the effects of craniofacial growth and surgical reconstructions. High-resolution CT has proved vital in determining the nature and extent of these syndromes. The radiologic evaluation of syndromes of the first and second BAs should begin first by studying a series of isolated defects: CL with or without CP, micrognathia, and EAC atresia, which compose the major features of these syndromes and allow more specific diagnosis. After discussion of these defects and the associated embryology, we proceed to discuss the VCFS, PRS, ACS, TCS, Stickler syndrome, and HFM.
Abbreviations
- ACS
- auriculocondylar syndrome
- BA
- branchial arch
- CL
- cleft lip
- CL/P
- cleft lip/palate
- CP
- cleft palate
- EAC
- external auditory canal
- HFM
- hemifacial microsomia
- MDCT
- multidetector CT
- PRS
- Pierre Robin sequence
- TCS
- Treacher Collins syndrome
- VCFS
- velocardiofacial syndrome
Radiographic evaluation of craniofacial deformities is necessary to define aberrant anatomy, plan surgical procedures, and evaluate the effects of craniofacial growth and surgical reconstructions.1 The recent rapid proliferation of MDCT is due, in part, to the increased utility of this technique for multiplanar bone and soft-tissue imaging. The definition of fine bony structure of the craniofacial anatomy on CT images is unmatched by other modalities. There has also been increased demand for treatment planning along with the advances in high-resolution CT evaluation and 3D reconstruction techniques.
Knowledge of the genetic basis of human disease and its effect on embryologic development has expanded greatly in recent years. Disorders of the first and second BA are generally thought to result from a combination of inadequate migration and inadequate formation of facial mesenchyma. Because many structures of the head and neck migrate during fetal development, an understanding of embryologic development helps determine the origin and nature of congenital lesions. Familiarity with craniofacial embryology and its associated effects on resultant anatomy also leads to a better understanding of the pathophysiologic basis of craniofacial syndromes. Additionally, it helps to establish a search pattern for characteristic radiologic features of many of these anomalies.
Part 1 of this review establishes the embryology, developmental anatomy, clinical symptoms, and characteristic imaging features of the isolated defects that compose some of the major features of the syndromes of the first and second BAs. Part 2 of this review discusses the syndromes and their radiographic features: PRS, HFM, ACS, TCS, Stickler syndrome, and VCFS. When applicable, the disorders number of the public data base of bibliographic information about human genes and genetic disorders—the Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/omim)—is given.
Imaging Approaches and Techniques
CT is the imaging technique of choice for studying syndromes of the first and second BAs. Modern MDCT scanners offer the additional ability to reconstruct facial bone data for dedicated evaluation of the middle and inner ear. We recommend acquiring images in the axial plane and reformatting sagittal and coronal high-resolution CT scans through the midface in planes parallel and perpendicular to the hard palate. Images should be obtained axially with a bone algorithm at a section thickness and interval of ≤3.0 mm. We recommend a 1.5-mm section thickness obtained by using a helical technique with 50% overlap of sections. Intravenous contrast may be administered in surgical planning to evaluate the aberrant course of the internal carotid artery but is typically not necessary. 3D CT reconstruction often allows a unified appreciation of abnormalities, which may aid in the detection of abnormalities and formulation of differential diagnoses. The complex interrelationship of malformations seen in craniofacial syndromes is often not adequately conveyed on axial and nonaxial planar reformations.1 3D imaging provides clinicians, radiologists, and patients with a quick easy-to-understand overview of craniofacial structures. These 3D representations can also be used for life-size model formation that can be used in surgical planning.
Embryology of the First and Second BAs and Associated Structures
Development of the craniofacial structures is a complex process that proceeds in an orderly fashion throughout embryonic and fetal stages of formation. Craniofacial growth occurs due to a relatively rapid and orderly composition of mesodermal and cranial neural crest cells via a complex signaling network. Syndromes of the first and second BAs manifest along a spectrum of hypoplasia and aplasia of the structures composing these arches. Some differences between abnormalities of the first and second BA derivatives may reflect differences in the embryologic age at the time of the insult with respect to neural crest cell migration. Other changes are related to deregulation of cell-signaling pathways triggered by a combination of genetic and environmental factors.2 The manifestation and severity of the congenital abnormality depend on the alteration of gene-expression profiles.2,3 The pluripotent nature of synchronously migrating cells is thought to, at least partially, explain the appearance and pattern of mesenchymal and epithelial abnormalities seen with syndromic defects of the BAs. Multiple craniofacial syndromes have been shown to result from an abnormality in the quantity or quality of neural crest cell migration (ie, TCS and VCFS).4,5
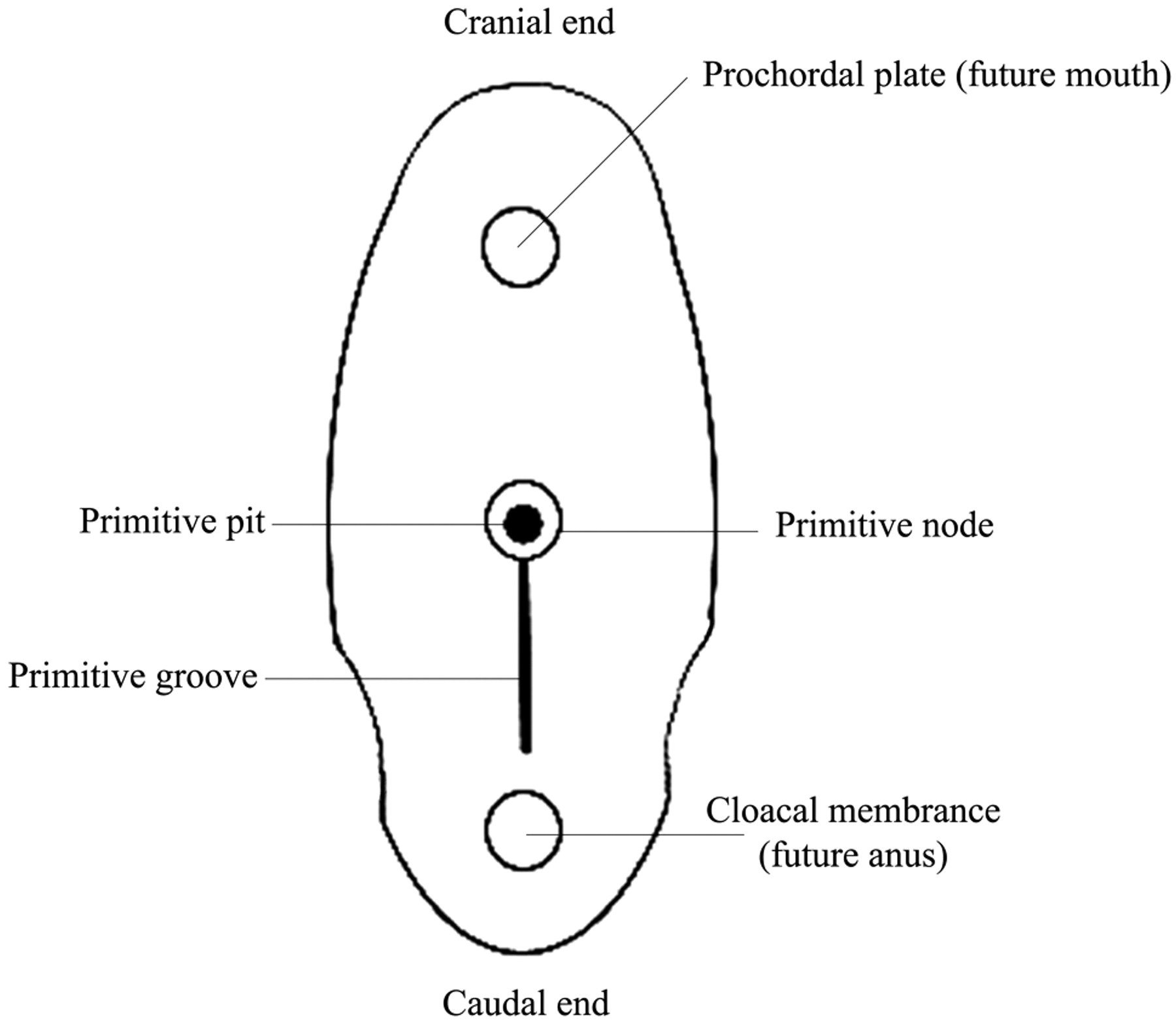
The 3 primary germ layers, ectoderm, mesoderm, and endoderm, form on the embryonic trilaminar germ disk and are the basis of all tissue and organ formation. The prechordal plate at the cranial end and the cloacal plate at the caudal end characterize the embryonic poles of the germ disk, which form due to opposing zones of deficient mesoderm (Fig 1). The prechordal plate is formed by the sinking inward of the oropharyngeal membrane. This creates a central depression for a key central structure in the formation of the face, the stomodeum. The frontal prominence develops superior to the stomodeum during the fourth postovulatory week and gives rise to the superior and middle portions of the face, comprising the area between the upper lip and forehead.6,7 The maxillary and nasal swellings form beneath the frontal prominence. Synchronously with the formation of the nasofrontal prominence, there is formation of 6 mesodermal arches that are separated from each other externally by ectodermally lined branchial clefts (grooves) and internally by endodermally lined pharyngeal pouches (Fig 2).8

Dorsal aspect of the germ disk from an approximately 15-day embryo.
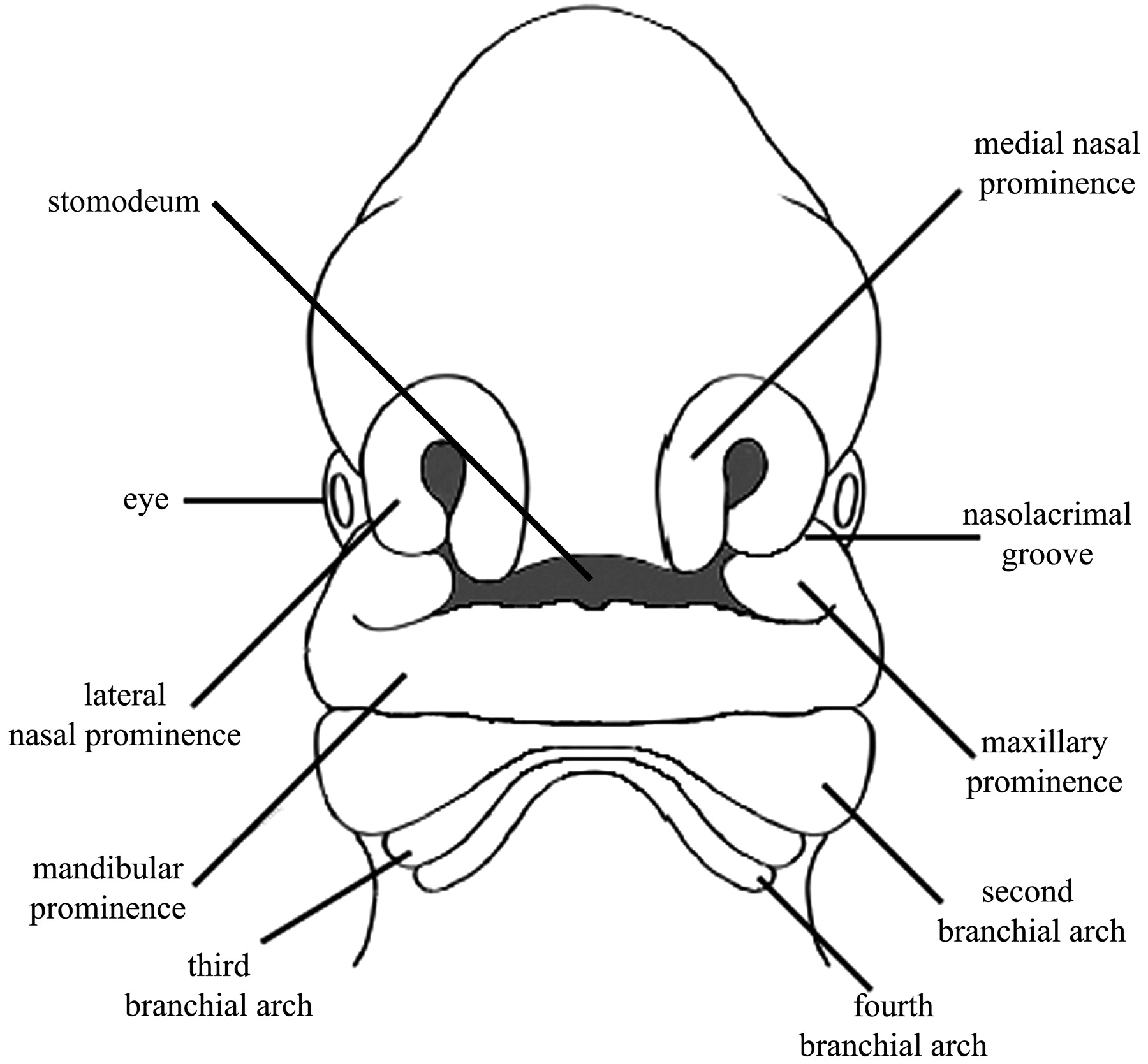
Frontal view of an approximately 30-day embryo showing the positions of the stomodeum relative to the medial and lateral nasal prominence and the maxillary and mandibular prominences.
Although development of pharyngeal arches, clefts, and pouches resembles formation of gills in fish and amphibia, in the human embryo, real gills (branchia) are never formed. The term pharyngeal has been alternatively offered for use in the human embryo; however, “branchial” continues to be the more commonplace term and thus is used in this article.9 The BAs have a significant impact on the external appearance of vertebrates. Originally, they consist of slabs of mesenchymal tissue divided by the branchial clefts. At the end of the fourth week of gestation, 4 well-defined pairs of BAs contribute to the characteristic external appearance of the human embryo.7,10
The mandibular prominence of the first arch lies caudal to the stomodeum. The maxillary prominence represents the dorsal portion of the first BA and is located lateral to the stomodeum and the frontonasal prominence. The mesenchyme of the maxillary process gives rise to the maxilla, zygomatic bone, and a part of the temporal bone through membranous ossification. The mandible is also formed by membranous ossification of mesenchymal tissue surrounding the Meckel cartilage, the cartilaginous mesenchymal component of the first BA. The first BA additionally gives rise to the muscles of mastication, the short crus and body of the incus and the head of the malleus, parts of the auricle, the anterior two-thirds of the tongue, and the mandibular branch of the trigeminal nerve.
The second or hyoid arch enlarges and grows so that by the sixth week, it will overlap and cover the third, fourth, and sixth arches. The Reichart cartilage is the mesenchymal contribution to the second arch that forms the styloid ligament; the manubrium of the malleus; the long process of the incus; the head, neck, and the crura of the stapes; and portions of the body and the lesser horn of the hyoid bone. The second arch also contributes the muscles of facial expression, the stapedius, the stylohyoid, and the posterior belly of the digastric muscle. These muscles are innervated by the facial nerve, though they migrate into the territory of the first BA.10–13
Specific neural crest cell segregation is critical to prevent fusions of the ectodermal and mesenchymal elements and also to prevent mixing of neural crest cells with different genetic constitutions.3 This migrational isolation leads each pharyngeal arch to consist of a core of specific mesenchymal tissue covered on the outside by surface ectoderm and on the inside by epithelium of endodermal origin. The core of each arch comprises neural crest cells that migrate along the BAs, helping to form the characteristic muscular, cranial nerve, and arterial component of each arch (Table 1).7,10,14,15
Derivatives of the BAs
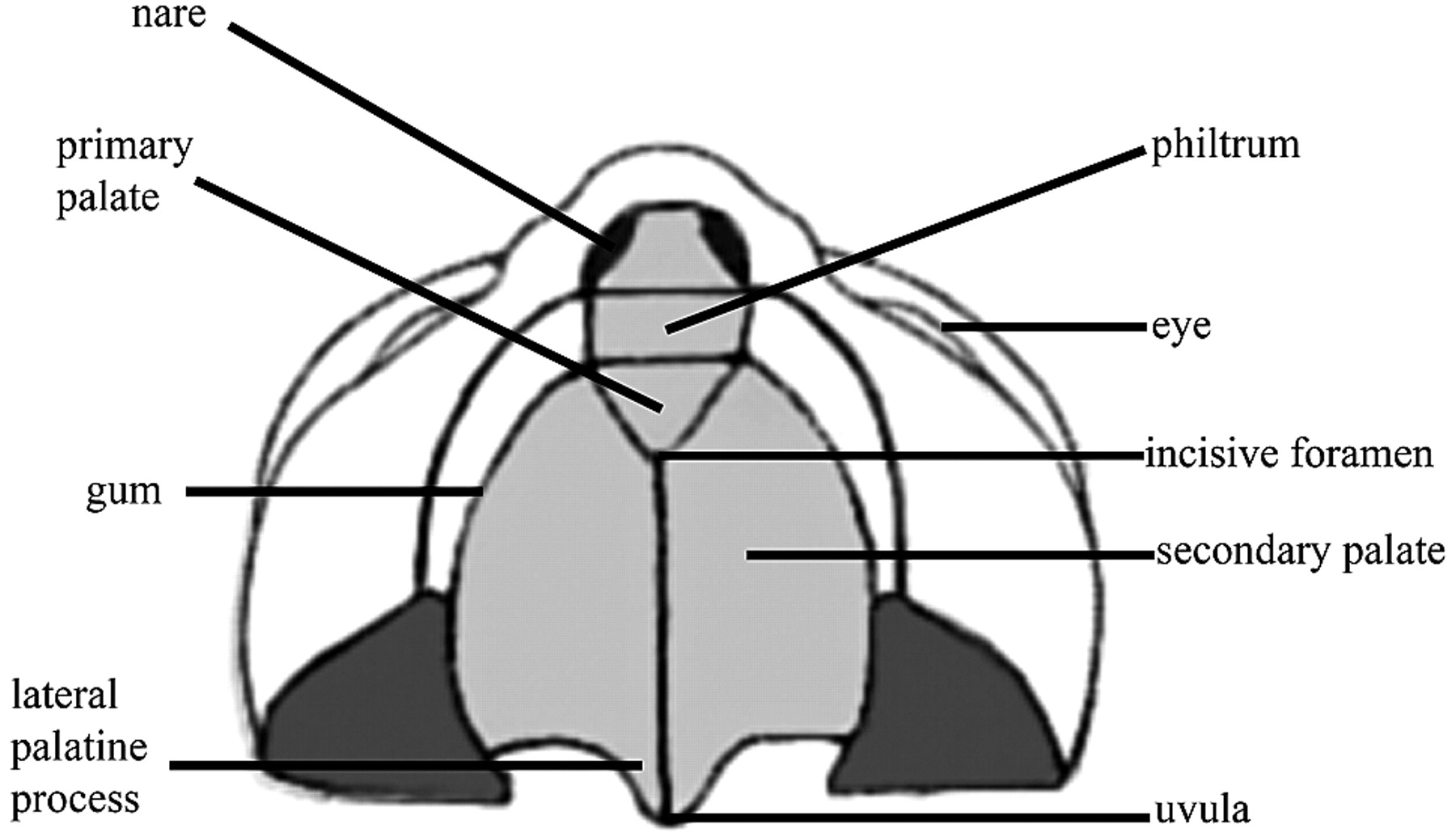
The philtrum and primary palate, the structures anterior to the incisive foramen, begin to form at approximately 5 weeks of gestational age by the coalition, growth, and differentiation of the frontonasal process and the fusion of the 2 medial nasal prominences. The fusion of medial nasal prominences gives rise to the intermaxillary segment of the frontonasal process. This structure is the origin of the philtrum and the portion of the maxilla from which the incisors arise. During the fifth and sixth gestational weeks, medial growth of the maxillary prominences results in fusion of the medial nasal and maxillary prominences. This leads to formation of the upper lip and anterior alveolus (Fig 3). The most common type of cleft lip results from failure of the maxillary swellings to fuse with the intermaxillary process.7,16–18
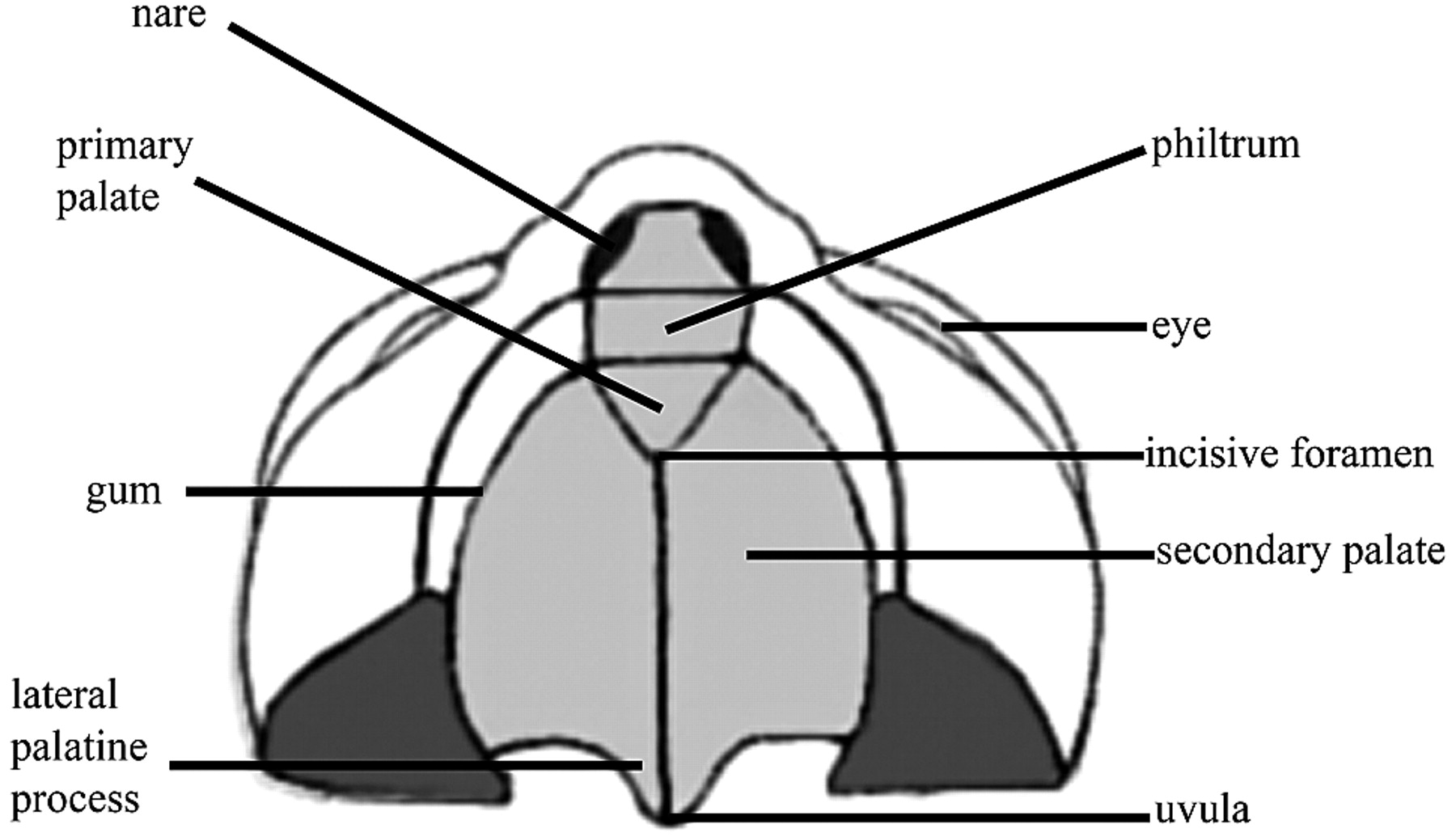
Ventral illustration of the palate, incisive foramen, gum, lip, and nose.
Formation of the secondary palate parallels that of the primary palate. The secondary palate, the portion posterior to the incisive foramen, forms through the fusion of paired outgrowths of the maxillary prominences, the palatal shelves. The shelves appear during the sixth week of development as vertical projections into the oral cavity on the lateral aspects of the tongue. In the seventh week of gestational development, the shelves elevate to a horizontal orientation and fuse, closing the secondary palate. Fusion begins anteriorly at the incisive foramen and proceeds posteriorly to completion around the 12th gestational week. Failure of complete closure of this process or complete elevation of the palatal shelves leads to CP. The tongue does not directly participate in palate closure; however, altered tongue position or function may mechanically block fusion of the palatal shelves, as seen in PRS, and can occur in cases of severe micrognathia and syndromes associated with poor neuromuscular control (ie, Trisomy 21).19–22
Isolated Defects
The radiologic evaluation of syndromes of the first and second BAs should begin with studying a series of isolated defects that compose some of the major features of these syndromes and allow a more specific diagnosis.
Facial Clefting
Facial clefting, CL with or without CP, is a common congenital malformation, accounting for 13% of all congenital anomalies, second only to clubfoot as the most frequent major birth anomaly.23 It is the most common congenital craniofacial malformation.24 Prevalence of CL/P averages approximately 1 in 700, with variations among races and between genders.25 Although in many neonates, CL/P is isolated, 29% may be associated with an underlying disorder.26,27 CL/P is associated with >300 syndromes including ACS, TCS, PRS, Goldenhar syndrome, Stickler syndrome, and VCFS.16,27,28
Facial clefting has a major clinical impact, requiring surgical, dental, orthodontic, speech, hearing, and psychological management throughout childhood. The etiology of CL/P is mostly unknown, but both genetic and environmental factors play a role.29 There is marked racial and geographic variability observed, with a higher prevalence seen among Native Americans (3.6 per 1000) and a lower frequency among African Americans (0.5 per 1000). CL/P is etiologically distinct from CP alone. First-degree relatives of patients with CL/P have an increased incidence of CL/P but not of CP alone. Relatives of patients with CP alone have an increased frequency of CP but not of CL/P.30
Failure of fusion between any of the facial structures (eg, failure of maxillary swellings to fuse with the intermaxillary process leading to cleft; see discussion above) results in a cleft, which may be unilateral or bilateral. Clefting of the lip and palate is seen along a spectrum, extending from occult discontinuities within the orbicularis oris muscle, which may be detected by using high-resolution postnatal sonography,22 to grossly visible clefts involving skin, muscle, and bone.
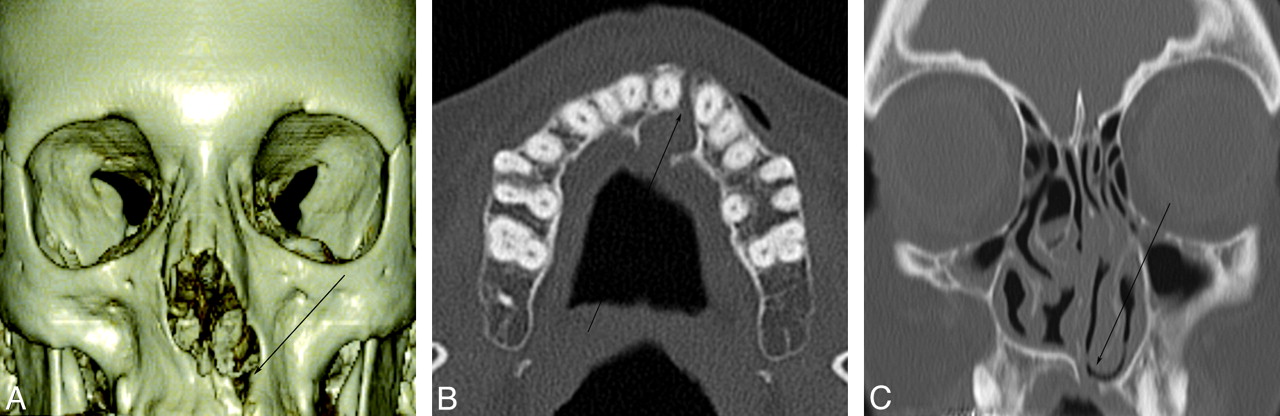
Radiologic evaluation of facial clefting should focus on searching for primary defects of the lips and palate and then proceed in a systematic fashion to associated defects (Fig 4). Sonography can be used to identify clefting prenatally in the lip and primary palate (anterior alveolar ridge). Prenatally identifying a cleft in the secondary palate or an isolated cleft palate is difficult and virtually impossible with older equipment. Thus, the role of careful postnatal clinical evaluation remains vital. CT evaluation of facial clefting is typically reserved for complex cases and those with defects or complications outside the lip and palate.

A, A 44-year-old woman with CP. 3D bony reconstruction shows a bony cleft (arrow) extending from the left aspect of an asymmetrically enlarged pyriform aperture to the alveolar surface. B, Axial CT image shows a bony cleft (arrow) between the left central and lateral maxillary incisors. C, Coronal CT image shows the extension of the bony clefting (arrow) to involve the primary palate.
Auricular Atresia
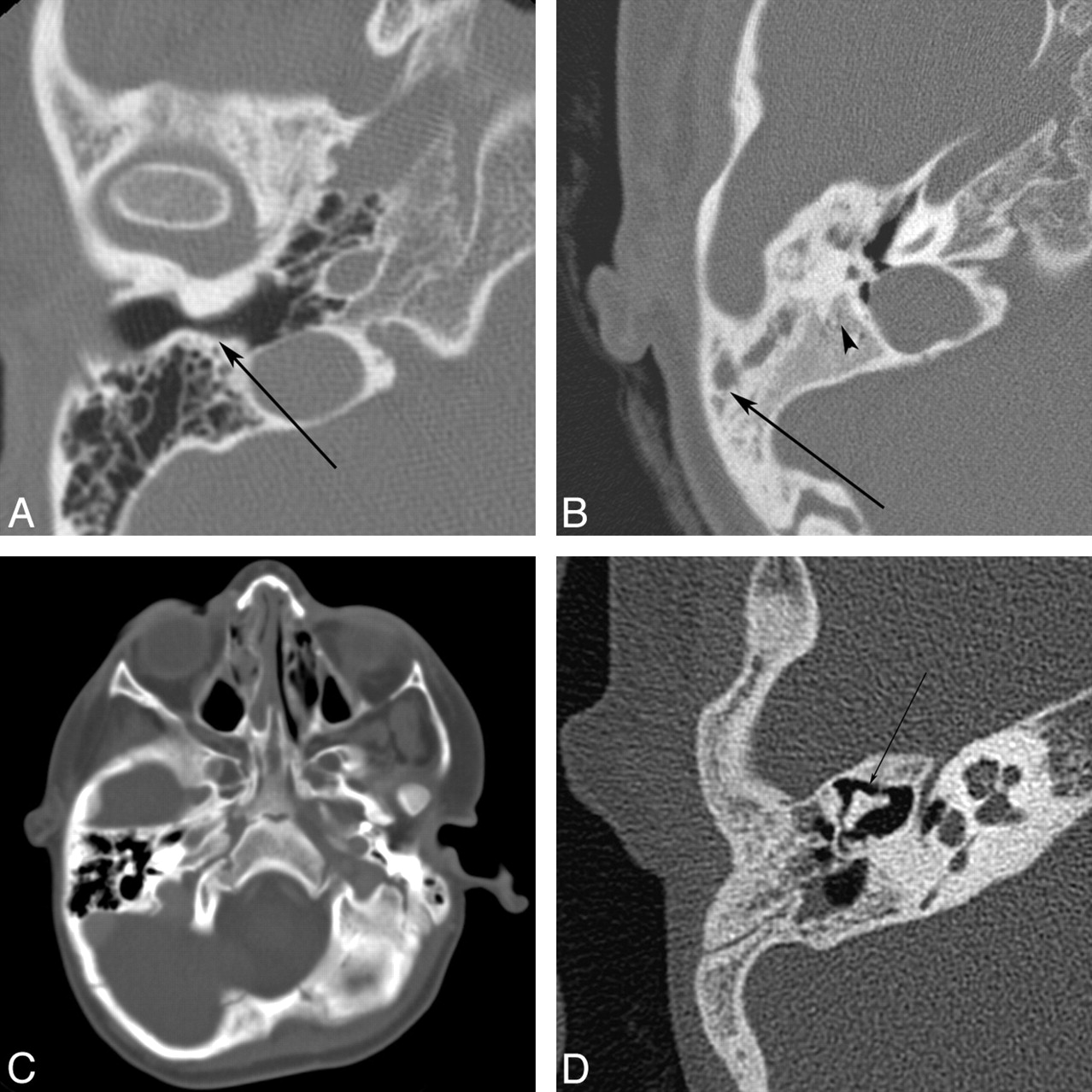
Auricular atresia occurs along a spectrum from an isolated malformed auricle to, at the most extreme form, an absent EAC with severe inner, middle, and external ear defects. These severe cases of EAC atresia are sometimes associated with a bony plate that replaces the tympanic ring and forms the lateral wall of the dysplastic middle ear cavity. Due to the common embryologic origin, EAC abnormalities are often associated with abnormalities of the external and middle ear. Middle ear defects can be subtle or severe and include absent or maldevelopment of any of the ossicles, with alteration of other structures of common embryologic origin (such as the course of the facial nerve) (Fig 5).12,31–33
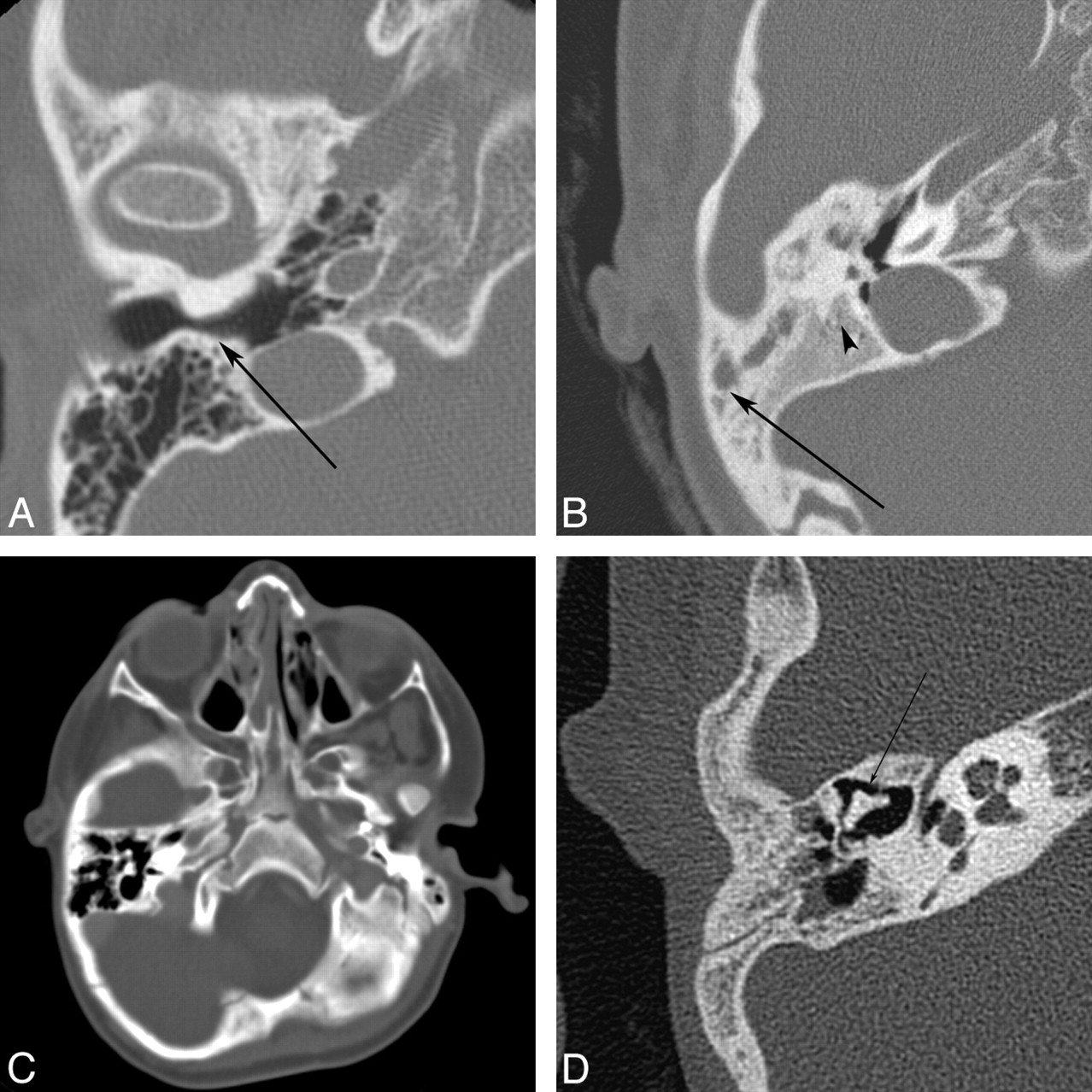
Auricular atresia in various degrees of severity. A, Axial CT image in a 64-year-old woman with nonsyndromic EAC atresia shows marked narrowing of the bony EAC (arrow). B, Axial CT image in a 9-year-old girl shows severe atresia with a lateral bony plate (arrow). The middle ear cavity is small and dysplastic (arrowhead). There is also ipsilateral microtia. C, Axial CT image in a 3-year-old boy with Goldenhar syndrome shows complete bony atresia of the right EAC. D, Axial CT image in a 6-year-old boy with unilateral auricular atresia with associated ossicular chain fusion (arrow) and microtia (not shown).
Jahrsdoerfer et al34 described a 10-point rating scale for the selection of surgical candidates by comparing high-resolution CT findings with postsurgical hearing results. Using 9 reproducible criteria, one calculates a score to predict postoperative improvement of the speech-reception threshold (Table 2). The criteria include assessment of the stapes, oval window, round window, middle ear space, mastoid pneumatization, facial nerve course, malleus-incus complex, and incus-stapes articulation. One point is given for each item with a normal or slightly dysplastic appearance. The stapes is an exception, for which 2 points are given when present. The final point is based on the clinical appearance of a fairly developed auricle. Patients with ≥6 points on the grading system are considered possible candidates for surgical reconstruction. This method has demonstrated clinical utility and also provides a useful evaluation system for the radiologist and otologist. The proper use of this rating scale relies on the availability of high-quality CT examinations and radiologists with detailed knowledge of the relevant anatomy.
System of Jahrsdoerfer et al for preoperative evaluation of aural atresia and stenosis as assessed using high-resolution CT of the temporal bonea
Auricular atresia can produce a number of problems for patients, including audiologic, cosmetic, and other associated clinical problems.12 EAC atresia has been described as part of various syndromes including TCS, ACS, PRS as well as Goldenhar, Crouzon, Möbius, Klippel-Feil, Fanconi, VCFS, Vater, and CHARGE syndromes.35
Micrognathia
Micrognathia is a frequently encountered facial abnormality in which the mandible is hypoplastic. Micrognathia is largely associated with craniofacial syndromes; however, nonsyndromic cases do arise. A study by Singh and Bartlett36 showed that of 266 patients with micrognathia, only 18 had congenital micrognathia without an identified syndrome. Micrognathia can be accompanied by the full spectrum of PRS (micrognathia, cleft palate, and relative macroglossia) and is also a dominant feature in ACS, HFM, TCS, and Stickler syndrome. It may also be seen variably in VCFS.17
Milder forms of micrognathia are common in infants and typically resolve with growth of the mandible. Radiologic evaluation of micrognathia should focus on the degree of mandibular hypoplasia, the temporomandibular joint, and the condyle and coronoid processes. One should also look for the often-associated abnormalities of the auricle, maxilla, and palate. Micrognathia is typically associated with malocclusion (abnormal tooth alignment), which may require orthodontic treatments and/ or tooth extraction. Preoperative CT evaluation is important for surgical planning and postoperative assessment of improvement (Fig 6).

A 6-year-old boy with syndromic micrognathia. A−C, 3D bony reconstructions show mandibular hypoplasia and abnormal temporomandibular joints, condyles, and coronoid processes. D, Axial CT image shows severe micrognathia and malocclusion.
In syndromes involving micrognathia, the oromandibular abnormalities often require the most intensive medical intervention. In severe cases of mandibular hypoplasia, glossoptosis may lead to upper respiratory tract obstruction, with mortality as high as 30%, due to the combined effects of malnutrition, airway obstruction, and failure to thrive.37,38 General anesthesia can be problematic due to problems with intubation. Glossoptosis is also associated with snoring, apnea, and sleep disturbance. In less severe cases, malocclusion may lead to masticatory abnormalities that require orthodontic treatment or orthognathic surgery. Speech therapy may also be required to treat the articulation defects that may be seen in some patients. Microsomia and limited mandibular excursion can produce difficulty with intraoral examinations and treatments that require intraoral manipulation.
Conclusions
The first and second BAs are the embryologic origin of many of the structures of the face. A wide variety of congenital conditions may arise from their contents. A knowledge of the anatomic formation of this region is important in understanding abnormalities in development, which in turn aids in the formulation of precise diagnoses and differential diagnostic considerations.
Acknowledgments
We thank Michael Cunningham, MD, PhD, for his assistance with manuscript preparation.
Indicates open access to non-subscribers at www.ajnr.org
References
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Craniofacial features of POLR3-related leukodystrophy caused by biallelic variants in POLR3A, POLR3B and POLR1C
- Craniofacial features of POLR3-related leukodystrophy caused by biallelic variants in POLR3A, POLR3B and POLR1C
- Loss of function of the E3 ubiquitin-protein ligase UBE3B causes Kaufman oculocerebrofacial syndrome
- Bilateral Preauricular Pits