
Abstract
SUMMARY: We describe how proton MR spectroscopy (1H-MR spectroscopy) was useful in elucidating the diagnosis of galactosemia in an undiagnosed 6-month-old infant. In vivo 1H-MR spectroscopy of the brain showed a doublet at 3.7 parts per million, which was identified as galactitol (Gal-ol) by in vitro 1H-MR spectroscopy of the urine. Galactosemia was subsequently confirmed by laboratory tests and treatment was initiated. A follow-up brain MR imaging and 1H-MR spectroscopy study revealed resolution of white matter lesions and disappearance of Gal-ol peaks.
Inborn errors of metabolism can lead to accumulation of metabolites in the brain and consequent abnormal peaks in the proton MR (1H-MR) spectroscopy spectrum. Patients with disorders in the carbohydrate metabolism can show abnormal brain 1H-MR spectroscopy with unusual peaks in the region of the sugars, around 3.7 ppm. It was shown that in vitro 1H-MR spectroscopy of the urine can help in the correct assignment of these peaks.1 We report here a case where in vivo and in vitro 1H-MR spectroscopy aid in the diagnosis of galactosemia.
Case Report
A 6-month-old female patient presenting with delay of psychomotor development, bilateral ocular cataracts, hepatomegaly, and hypoattenuation of the brain white matter on CT scan was sent to our department for MR imaging and 1H-MR spectroscopy examination to elucidate her diagnosis.
MR imaging showed diffuse and symmetrical involvement of the frontal, temporal, parietal, and occipital white matter, characterized by hypointensity on T1- and hyperintensity on T2 -weighted images, extending to “U” fibers (Fig 1A). In the temporal, frontal, and parietal regions hypointense areas associated with hyperintense white matter involvement on fluid-attenuated inversion recovery (FLAIR) images could be observed (Fig 1B). There was no gadolinium enhancement. Corpus callosum, internal and external capsules, and optic radiations were spared. The diffusion-weighted images (DWIs) showed hypointense lesions in the areas corresponding to white matter involvement with increased apparent diffusion coefficient (ADC) values (Fig 1C).

Images obtained before dietary treatment. Axial T2-weighted image (A) showing hyperintense bilateral diffuse white matter involvement reaching the “U” fibers. The internal capsules are spared. The lesions are hyperintense with hypointense areas on FLAIR images (B). ADC map (C) shows increased diffusion in the white matter lesions.
In vivo 1H-MR spectroscopy of the parieto-occipital white matter (Fig 2A) revealed normal NAA: N-acetylaspartate (NAA)/creatine (Cr) and choline (Cho)/Cr, but reduced myo-inositol (mI)/Cr when compared with control values (Table 1). Unknown signals of very high concentration were found in the region of 3.5 to 4.0 ppm (Fig 2B). These signals were partially inverted when the spectrum was acquired with a TE of 135 milliseconds. This behavior is characteristic for carbohydrate signals. For a more detailed study of the carbohydrate composition, an in vitro 1H-MR spectroscopy of the urine was acquired by using a high-resolution 500-MHz spectrometer, and the result was compared with the urine of a normal volunteer. We could observe a group of multiple peaks in the region of 3.6 to 3.9 ppm (Fig 3) and a triplet at 3.98 ppm, which were absent in the normal spectrum. We found also 2 clearly resolved doublets at 4.57 and 5.25 ppm in the patient’s urine, which were absent in the normal spectrum. The in vitro urine spectrum suggested the presence of high concentrations of galactose (Gal-ose) and galactitol (Gal-ol) in the patient’s urine. The diagnosis of galactosemia was later confirmed by the finding of low levels of galactose-1-phosphate uridyl transferase in the erythrocyte. Subsequently, the patient started a restricted lactose-free diet. At the age of 2 years, the follow-up MR imaging of the patient showed marked atrophy, more evident in the frontal lobes with enlarged sulci and dilation of the lateral ventricles more prominent in the anterior horns. There was marked improvement of the lesions with almost complete resolution of white matter signal intensity abnormalities. The sparse residual lesions were located in the basal temporal lobes and periventricular frontal regions, presenting hypointensity on T1-weighted and hyperintensity on T2-weighted (Fig 4) and FLAIR images, without gadolinium enhancement. On DWI, the lesions presenting increased ADC values on the previous examination had resolved. In vivo 1H-MR spectroscopy of the parieto-occipital white matter did not show evidence of Gal-ol signal intensity (Fig 5). Metabolite ratios appeared to be normal when compared with control group (Table).

Axial localizer T2-weighted image showing the MR spectroscopy voxel location (A). STEAM (TE/TR, 30/1500 milliseconds) (B) in vivo 1H-MR spectroscopy spectrum of the patient before treatment.

In vitro 1H-MR spectroscopy spectrum of the urine sample, showing the region of 3.2–4.1 ppm in the patient.
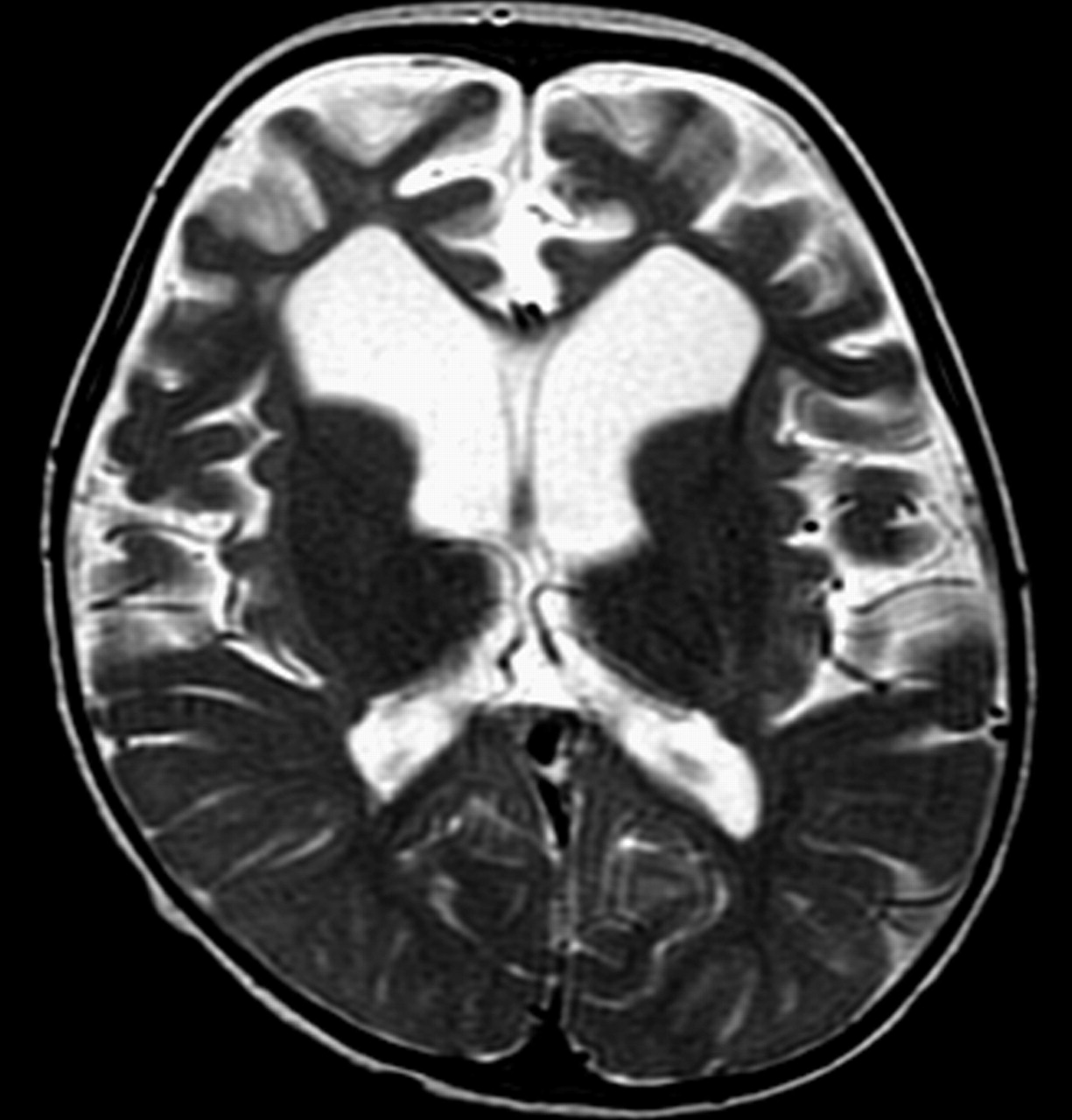
Images obtained after dietary treatment. Axial T2-weighted image shows marked frontal atrophy with enlarged sulci and ventricular dilation. Note also enlargement of Sylvian fissures and improvement of white matter lesions.

STEAM (TE/TR, 30/1500 milliseconds) in vivo 1H-MR spectroscopy spectrum of the patient after treatment.
| NAA/Cr | Cho/Cr | mI/Cr | Gal-ol/Cr | |
|---|---|---|---|---|
| Patient before treatment | 1.45 | 1.04 | 0.19 | 14.30 |
| Patient after treatment | 1.57 | 0.87 | 0.50 | |
| Controls* | 1.53 ± 0.22 | 0.89 ± 0.14 | 0.48 ± 0.07 |
Note:—Control mean values are included for comparison purposes. Cho/Cr indicates choline/creatine; Gal-ol/Cr, galactitol/creatine; ml/Cr, myo-inositol/creatine; NAA/Cr, N-acetylaspartate/creatine.
* Control group consisted of 10 healthy volunteers (3 boys and 7 girls) of mean age of 6 ± 1 years (range, 4–9 years).
Metabolite ratios in the parieto-occipital white matter measured by the STEAM technique (TE/TR = 30/1500 ms) before and after patient’s treatment
Discussion
Galactosemia is a disorder caused by a deficiency of any of the 3 possible enzymes involved in the metabolism of galactose: galactokinase, transferase, or epimerase. Any single deficient enzyme can result in accumulation of Gal-ol in the lenses, causing cataracts,2,3 and in the brain, leading to cerebral edema, probably due to an osmotic effect.2 The most common type of galactosemia, known as classic galactosemia, is caused by the deficiency of galactose 1-phosphate uridyltransferase (GALT). In humans, GALT deficiency leads to significant neonatal morbidity and mortality, which depends on Gal-ose ingestion, as well as long-term complications of primary ovarian failure and cognitive dysfunction, which are independent of diet.4 Although potentially lethal, neonate screening and Gal-ose restriction prevent neonatal hepatotoxic syndrome, but total exclusion of Gal-ose from the diet does not ensure the absence of all symptoms.5,6 Two different metabolites are potentially toxic: Gal-ol is responsible for the cataracts, whereas galactose-1-phosphate causes the rest of the clinical symptoms. Through measurement of urine and plasma Gal-ol levels, it is possible to distinguish galactosemic patients from normal subjects.7
Patients with galactosemia have been previously studied by MR imaging8 and 1H-MR spectroscopy.2,9,10 The MR imaging of galactosemic patients can present cerebral and cerebellar atrophy and multiple small hyperintense lesions in the white matter on T2-weighted images.8 Berry et al reported the first in vivo 1H-MR spectroscopy evidence of Gal-ol accumulation in a galactosemic patient in 2001.2 Gal-ol appears in the 1.5T 1H- spectrum as 2 peaks at 3.67 and 3.74 ppm, which can be observed in several locations of the brain. To be able to detect Gal-ol in the in vivo brain 1H-MR spectroscopy examination high Gal-ol levels still need to be present in the patient’s urine.9 Galactosemic patients, who have been following a Gal-ose-restricted diet for several years, and therefore present controlled levels of Gal-ol in the urine, do not present Gal-ol in the brain by in vivo 1H-MR spectroscopy.9
In our case, the patient performed the first MR imaging and 1H-MR spectroscopy examination without having a diagnosis, and we described how it was possible to aid in the diagnosis by performing in vivo and in vitro 1H-MR spectroscopy. Furthermore, we also showed how the 1H-MR spectroscopy reversed to normal once the patient was under dietary treatment.
The unusual peaks encountered around 3.7 ppm in the brain 1H-MR spectroscopy of our patient suggested the accumulation of carbohydrates, probably due to a failure in the patient’s carbohydrate metabolism. There are several metabolic diseases, which could lead to elevation of carbohydrates in the spectrum, like glucose in diabetes mellitus, Gal-ol in galactosemia, and arabitol, and ribitol in a recently reported inborn error of the polyol metabolism.1,11 All of these sugars present multiple peaks in the region of 3.7 ppm. To distinguish between these conditions by 1H-MR spectroscopy it is necessary to work with a higher spectral resolution than the one obtained in vivo at 1.5T. For this reason an in vitro analysis of the urine sample was performed. Moolenaar et al1 acquired in vitro urine spectra from a patient with an inborn error in the arabitol/ribitol metabolism, a patient with diabetes mellitus, and an untreated galactosemic patient. By comparing the latter one with the urine spectrum of our patient, we could observe many similarities in the region of 3.4–4.0 ppm, including the presence of the characteristic Gal-ol triplet at 3.98 ppm.11 It is difficult to correlate the peaks around 3.7 to a specific molecule, because they are multiple, and several of the sugars and polyols, like Gal-ose, Gal-ol, and galactonate resonate in this region, leading to an overlap of different peaks. Gal-ose presents doublets at 4.57 and 5.25 ppm.9 We could observe the presence of these doublet peaks in the in vitro spectrum, also confirming high levels of Gal-ose in the urine of our patient. In cases of GALT deficiency, initially there is an accumulation of Gal-ose, which is then reduced to Gal-ol.9
The presence of Gal-ol in high concentrations in the urine of our patient explains the prominent doublet observed at 3.7 ppm in the in vivo spectrum of the brain.
Gal-ol peaks in this region show J-coupling effects, which explain the inversion of these peaks by using a TE of 135 ms. Polyols, such as Gal-ol, can easily penetrate the cellular membrane. With the increase in Gal-ol, we expect to see a decrease of mI, to compensate the osmotic pressure.1 This could explain the low mI/Cr ratio found in the parieto-occipital white matter of our patient before treatment (Table).
The MR imaging and 1H-MR spectroscopy results obtained at the age of 2 years are consistent with other reports of treated galactosemic patients: normal brain 1H-MR spectroscopy spectrum9 and atrophy and sparse residual lesions on the MR imaging.8
Conclusion
1H-MR spectroscopy was very useful in the diagnosis of this galactosemic patient. The detection of unknown peaks in the spectrum must be considered and investigated. The in vitro MR spectroscopy was a very important tool to aid in this investigation.
Acknowledgments
The MRUI software package was kindly provided by the participants of the EU Network programs: Human Capital and Mobility, CHRX-CT94–0432, and Training and Mobility of Researchers, ERB-FMRX-CT970160.
Footnotes
This work was presented in part at Sociedad IberoLatinoAmericano de Neuroradiologia (SILAN), XVth congress, June 23–27, 2003, Estoril, Portugal.
References
- Received September 28, 2004.
- Accepted after revision January 31, 2005.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}