
Abstract
BACKGROUND AND PURPOSE: Because of improved visualization of posterior fossa structures with MR imaging, cerebellar malformations are recognized with increasing frequency. Herein we attempt to describe and propose a rational classification of cerebellar malformations.
METHODS: MR images obtained in 70 patients with cerebellar malformations were retrospectively reviewed. The cerebellar malformations were initially divided into those with hypoplasia and those with dysplasia. They were then divided into focal and diffuse malformations. Finally, they were separated according to other features, such as brain stem involvement and cerebral involvement.
RESULTS: All patients with diffuse cerebellar dysplasia (muscular dystrophy [n = 10], cytomegalovirus [n = 6], lissencephaly [n = 3],) had abnormalities of the cerebrum. Patients with focal cerebellar dysplasia of the Joubert (n = 12) and rhombencephalosynapsis (n = 8) types had variable cerebral dysplasia. Patients with nonsyndromic focal cerebellar dysplasia (isolated focal cerebellar cortical dysplasia [n = 2], cerebellar heterotopia with cerebellar cortical dysplasia [n = 1], idiopathic diffuse cerebellar dysplasia [n = 1], Lhermitte-Duclos syndrome [n = 1]) and those with cerebellar hypoplasia (isolated cerebellar hypoplasia [n = 6], pontocerebellar hypoplasia type 1 [n = 1]) had normal cerebra. Patients with features of Dandy-Walker malformation (n = 19) had both hypoplasia and dysplasia of the cerebellum. No notable difference was found between the cerebella of patients with large fourth ventricle cysts (Dandy-Walker malformations) and those without large fourth ventricle cysts (isolated cerebellar hypoplasia). Therefore, the Dandy-Walker malformation seems to be heterogeneous.
CONCLUSION: Use of this classification system helps in the segregation and understanding of the relationship among cerebellar malformations. Although it will undoubtedly require revisions, this classification is a first step in combining imaging with molecular biology to facilitate understanding of cerebellar development and maldevelopment.
Although many classifications have been presented for cerebral hemispheric malformations, no classification of cerebellar malformations has been widely accepted. Many individual conditions, such as Dandy-Walker malformation, have been extensively addressed in the radiologic literature; however, ongoing controversy exists regarding the relationship of Dandy-Walker malformation to other posterior fossa cystic malformations (1–4), and Dandy-Walker malformations may occur with other cerebellar malformations (5). Other malformations of the cerebellum, such as the so-called “molar tooth” type of midbrain-hindbrain malformations (including Joubert syndrome [6, 7], Arima syndrome [8], and Senior-Löken syndrome [5, 9, 10]), and rhombencephalosynapsis (11) are less well described, and their relationship to other cerebellar malformations is unclear. Other cerebellar hemispheric (12) and vermian (13, 14) malformations are described but poorly understood, with little reference in the literature other than in occasional case reports. Thus, the prognosis of patients with many cerebellar malformations is uncertain. Finally, it is often difficult to be sure whether abnormal cerebella identified in children with signs and symptoms referable to the posterior fossa represent atrophy, hypoplasia, or malformation (15–18).
Much of the confusion surrounding cerebellar malformations stems from a limited understanding of cerebellar embryology, limited histologic studies of these disorders, and lack of a practical classification of cerebellar malformations. We herein report the results of a retrospective analysis of 70 cases of cerebellar anomalies and review the literature with the aim of producing an imaging-based classification.
Methods
Patient Characterisitics
MR images obtained in 70 patients with a diagnosis of some form of cerebellar malformation were retrospectively reviewed. The images came from our clinical practice and from referrals sent to our practice for consultation and spanned 15 years. The clinical information available for the patients was extremely variable, excellent in some but sketchy in others; because the information was so inconsistent and because this classification was based largely on morphology, clinical information was not used as a basis for classification.
Seventy patients were studied. Nineteen patients had posterior fossa CSF collections and associated vermian or holocerebellar hypoplasia (sometimes referred to as the Dandy-Walker continuum) (4); 12, molar tooth malformations (sometimes referred to as Joubert malformation) (19); eight, rhombencephalosynapsis (11, 20); six, diffuse cerebral and cerebellar malformation secondary to congenital cytomegalovirus infection; six, cerebellar hypoplasia; 10, congenital muscular dystrophy syndromes (four with Fukuyama congenital muscular dystrophies, two with Walker-Warburg syndrome, two with muscle-eye-brain disease, two with merosin-deficient congenital muscular dystrophy); three, lissencephaly and cerebellar dysplasia (one with a known reelin mutation, the other two having no mutation identified); two, focal cerebellar cortical dysplasia; one, diffuse cerebellar dysplasia of unknown cause; one, pontocerebellar hypoplasia; one, cerebellar heterotopia; and one, Lhermitte-Duclos (Table 1). The 39 male and 31 female patients ranged in age from 2 days to 47 years (mean age, 3.1 years) at the time they were studied. Patients with Chiari malformations were not included in this review. Chiari malformations are considered to be the result of a lack of expansion of the embryonic fourth ventricle, with consequent hypoplasia of the posterior fossa (21), or anomalies of the craniovertebral junction (22, 23), not malformations of the cerebellum itself. We also did not include any patients with so-called “inverse cerebellum” (24) or “tectocerebellar dysraphia” (25), because we agree with Chapman et al (26) that these are better considered as subsets of occipital cephalocele, rather than as primary cerebellar malformations. Finally, findings in patients with isolated inferior vermian hypoplasia were not included in the study, because this condition is not considered abnormal at our institution, and therefore, was not a part of our collection of cerebellar anomalies.
Cerebellar Malformations
Definition of Terms
The cerebellar malformations were initially divided into those with hypoplasia and those with dysplasia. Hypoplasia was diagnosed according to the concepts of cerebellar atrophy and hypoplasia, as defined by Barkovich (27). We use the term cerebellar atrophy if the cerebellum was small with shrunken folia and large cerebellar fissures or if it had been shown to undergo progressive volume loss; because atrophy is considered the result of progressive metabolic injury and this study deals with anomalies, patients with cerebellar atrophy were excluded from this study. The term cerebellar hypoplasia is used for patients with a small cerebellum that had fissures of normal size compared with the folia. For the purposes of this study, hypoplasia is considered a disorder of cerebellar formation and patients with the diagnosis of hypoplasia are included. A structure was considered dysplastic if disorganized development, such as abnormal folial pattern or presence of heterotopic nodules of gray matter, was evident. We did not use the concept of hypogenesis of the vermis (formation of superior vermis but not inferior vermis) (27), because we have been shown that it is impossible to differentiate a vermis in which inferior lobules are missing (hypogenetic) from those in which some of the middle lobules are missing (dysplastic). Next, the malformations were divided into those that were generalized (those that involved both cerebellar hemispheres and the vermis) and those that were focal (localized to either a single hemisphere or the vermis).
Imaging Techniques and Analysis
MR imaging examinations were performed using many different images at multiple institutions during a 15-year period. All included sagittal T1-weighted and axial T1- or T2-weighted images. Coronal images were obtained for 49 of the 70 patients. The thickness of the imaging sections ranged from 3 mm to 8 mm.
Morphologic features based on MR imaging findings were recorded for each case. Both authors reviewed all cases and independently recorded the extent of cerebellar hemispheric and vermian development; folial pattern of the hemispheres and vermis (ie, normal vs abnormal), characteristics (ie, dysplastic, hypoplastic, or absent) of the hemispheres and vermis, relative size of the fissures and folia, presence of a posterior fossa CSF collection, size of the posterior fossa (which was assessed subjectively), and other evidence of mass effect, such as elevation of the torcula and bony remodeling and brain stem development.
Assignment of Anomalies
Both authors reviewed all the cases independently and classified them on the basis of specific criteria in the preceding paragraphs, which were derived from the literature and agreed upon beforehand. If disagreement existed, the cases were reviewed together and discussed, and a consensus was reached regarding classification. The patients were initially classified into conventional categories (eg, Dandy-Walker continuum, molar tooth malformation), whenever possible. These categories were then assigned to the classification system shown in Table 2. Those anomalies that did not fit any classic category were assigned to the category in Table 2 that best matched the morphology of the cerebellum, as defined in the preceding paragraph. Those cases that did not clearly fit into a category and those that might fit into more than one category were discussed by the authors in an attempt to make a logical assignment and were then assigned to the agreed upon category. Those discussions are elaborated in subsequent sections.
Classification Scheme for Cerebellar Malformations
Results
Cerebellar Hypoplasia
Dandy-Walker Continuum
Nineteen patients had vermian hypoplasia, ranging from mild to severe, associated with a retrocerebellar CSF collection that appeared continuous with the fourth ventricle. These patients fit the definition of the Dandy-Walker continuum, as defined by Barkovich et al (4) and subsequently modified (2, 3). Of the 19 patients, 10 had associated moderate to severe cerebellar hemispheric hypoplasia. Eleven patients displayed clear evidence of an enlarged posterior fossa with elevation of the torcula and thus fit the criteria of the classic Dandy-Walker malformation. Of these, nine had severe vermian and hemispheric hypoplasia, whereas the other two had moderate vermian hypoplasia with mild hemispheric hypoplasia. Eight patients had large posterior fossa CSF collections without definite elevation of the torcula. Of these, three had severe vermian hypoplasia (including one with hemispheric hypoplasia), one had moderate vermian hypoplasia with normal cerebellar hemispheres, and one had what appeared to be vermian dysplasia (Fig 1).
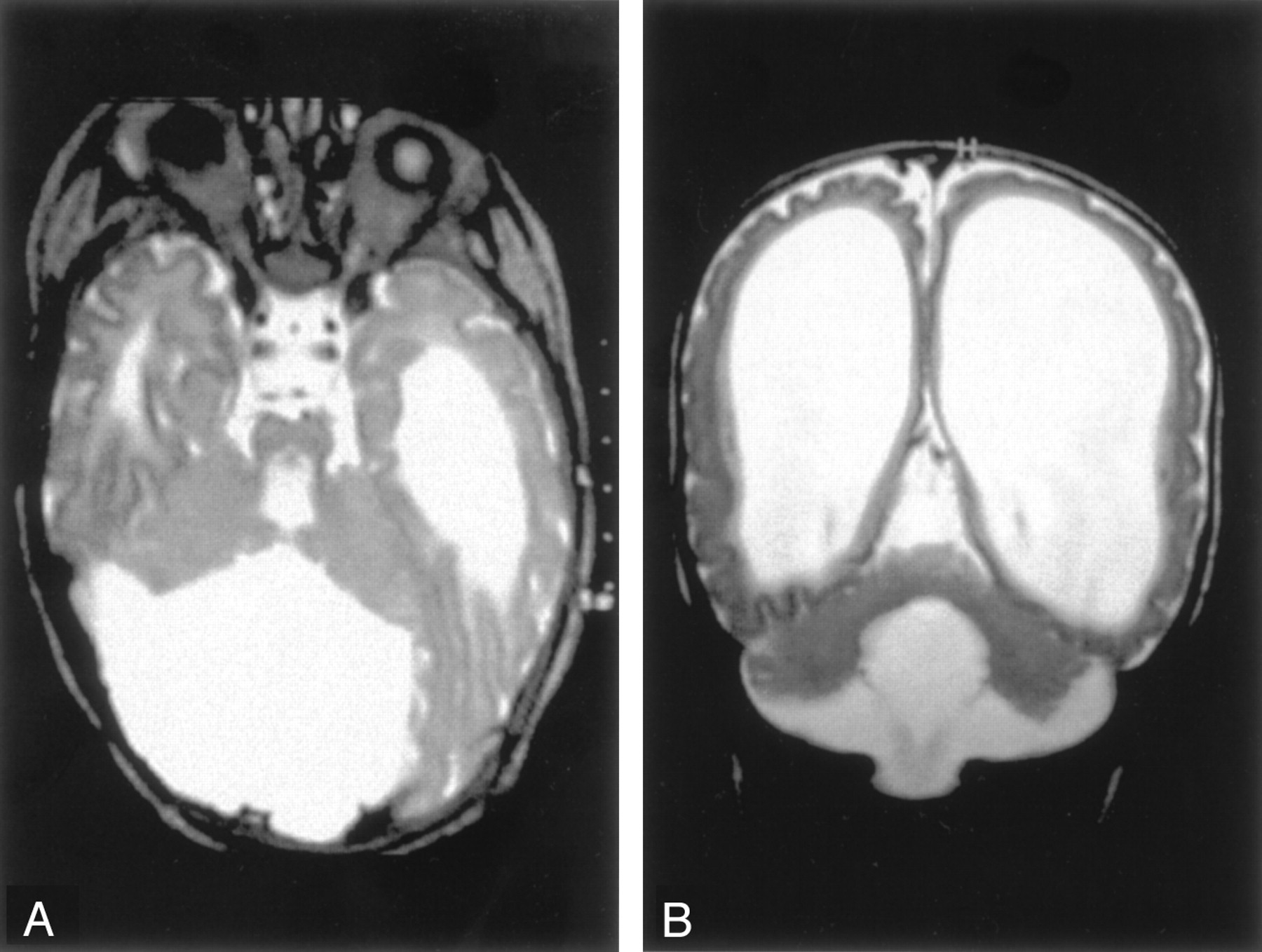
Dandy-Walker malformation with cerebellar dysplasia in a 4-month-old infant.
A, Coronal fast spin-echo (3000/102 [TR/TE]) image shows that the vermis is hypoplastic and the posterior fossa large, with the cerebellar hemispheres widely separated. The cerebellar hemispheres have an abnormal folial pattern, compatible with dysplasia. The cerebral hemispheres are abnormal with marked thinned white matter, enlarged ventricles, and shallow sulci.
B, Axial fast spin-echo (3500/112) image shows, in addition to the large posterior fossa fluid collection and dysplastic cerebellar hemispheres, an abnormality of the pons, which is small and has a ventral cleft.
Analysis of the cerebellum revealed that 16 patients had either isolated vermian hypoplasia or vermian hypoplasia in association with hypoplasia of the cerebellar hemispheres, whereas three had what appeared to be cerebellar dysplasia (the folial pattern was abnormal), involving both vermis and hemispheres (Figs 1 and 2). Of interest, the cerebral hemispheres were abnormal in two of these, with diminished volume of white matter and abnormal cerebral cortical gyral pattern (Fig 1). One case with vermian dysplasia had a small, bifid pons (Fig 1). When the hemispheres were hypoplastic or dysplastic, they were sometimes asymmetric but always bilaterally involved. Mass effect from the CSF collection was present in the patient with dysplasia, those with isolated vermian hypoplasia, and those with hemispheric involvement. Those cases without evidence of mass effect showed considerable morphologic overlap with cases of isolated cerebellar hypoplasia, whether focal or generalized.

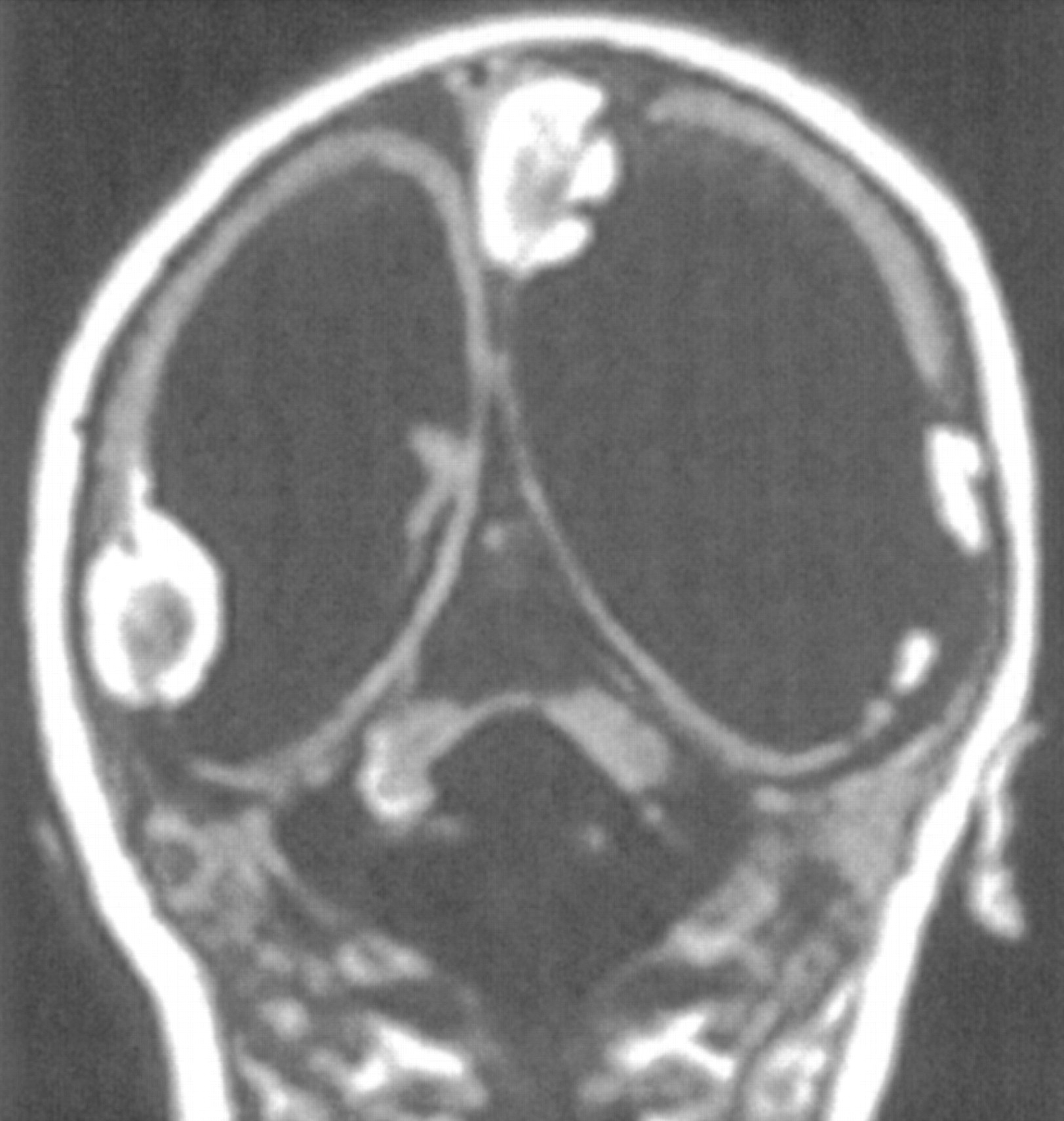
Dandy-Walker malformation with cerebellar dysplasia in a 6-day-old neonate.
A, Sagittal spin-echo (550/16) image shows large posterior fossa CSF collection and dysplastic appearing cerebellar vermis.
B, Axial spin-echo (3000/120) image shows abnormal folial pattern of cerebellar hemispheres.
Associated supratentorial anomalies included agenesis of the corpus callosum in three patients, hypogenesis of the corpus callosum in three patients, hydrocephalus in eight patients, and a single subependymal heterotopion in the left frontal lobe of one patient. Of interest, the patients with callosal anomalies all had classic Dandy-Walker malformations with severe posterior fossa enlargement; thus, six of 11 patients with classic Dandy-Walker malformations had callosal anomalies. In contrast, none of the eight patients with lesser degrees of posterior fossa enlargement had callosal anomalies.
Cerebellar Hypoplasia
Six patients had cerebellar hypoplasia. Of these, three had generalized hypoplasia of the cerebellar hemispheres and vermis, which was moderate in two patients and severe in one (Fig 3). The cerebral hemispheres were normal in these patients, but all had small brain stems, particularly the pons and medulla. All were adults who were referred for imaging for recent onset of neurologic problems (mild ataxia in two, change in character of headaches in one). These patients had no associated enlargement of the posterior fossa and no associated cyst. Again, no specific morphologic features distinguished this group from those cases of Dandy-Walker spectrum that did not have enlargement of the posterior fossa.

Cerebellar hypoplasia in a 33-year-old man. Sagittal spin-echo (500/11) image shows a profoundly small cerebellum in a fluid-filled, but normal-sized, posterior fossa. The pons and medulla are abnormally small.
We observed isolated unilateral hypoplasia of the cerebellum in two children (ages 1 and 5 years) and in one adult. The two children underwent imaging for developmental delay, whereas the adult was studied for persistent headaches. At examination, all patients exhibited mild ataxia but were otherwise neurologically normal. All had the appearance of a small hemisphere unilaterally despite normal-appearing vermis, brain stem, and contralateral hemisphere (Fig 4). In one patient, the contralateral hemisphere appeared slightly rotated. The cerebral hemispheres were normal in these patients.
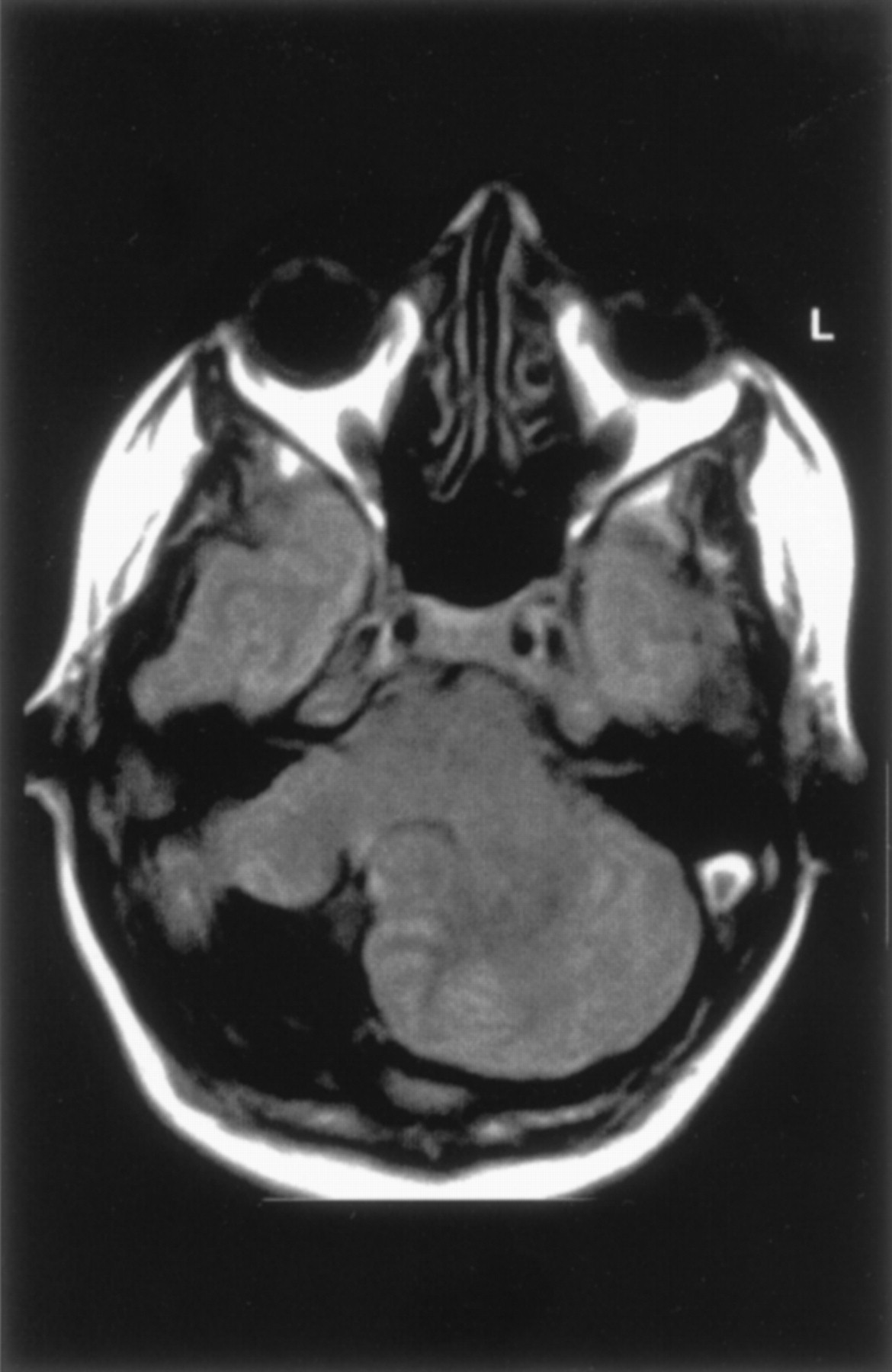
Unilateral cerebellar hypoplasia in 29-year-old woman. Axial spin-echo (2500/30) image shows a small right cerebellar hemisphere. The left hemisphere and vermis appear normal.
The patient with a clinical and radiologic diagnosis of pontocerebellar hypoplasia, type 1, revealed moderate hypoplasia of the cerebellar hemispheres, vermis, pons, and medulla. This condition was classified under generalized hypoplasia, because the small cerebellum had a normal folial pattern.
Cerebellar Dysplasia
Diffuse Dysplasias
Lissencephaly with Cerebellar Dysplasia.—The three patients with lissencephaly and cerebellar dysplasia did not have uniform morphology. The patient with reelin mutation had an unusual, smooth-appearing cerebellum. In addition to the small size of both the vermis and the hemispheres, no lobulation was observed; no folia and no fissures were present. Signal intensity of the cerebellum was normal. The other two patients had tiny cerebellar hemispheres and no visible vermis (Fig 5). Minimal foliation was observed; therefore, the malformations were classified as dysplasia. The brain stem (particularly the pons) was markedly small in all three patients. All three had nearly complete agyria in the cerebral hemispheres. The patient with the reelin mutation had hypogenesis of the corpus callosum, whereas the other two had complete callosal agenesis.

Lissencephaly and cerebellar hypoplasia in a 4-day-old neonate.
A, Sagittal spin-echo (550/11) image shows complete cerebral lissencephaly and a very small cerebellum. The brain stem is abnormally thin. The colliculi are fused.
B, Axial spin-echo (550/16) image shows the very small cerebellar hemispheres and the small pons with a central cleft, presumably due to absence of the crossing ventral pontine axons.
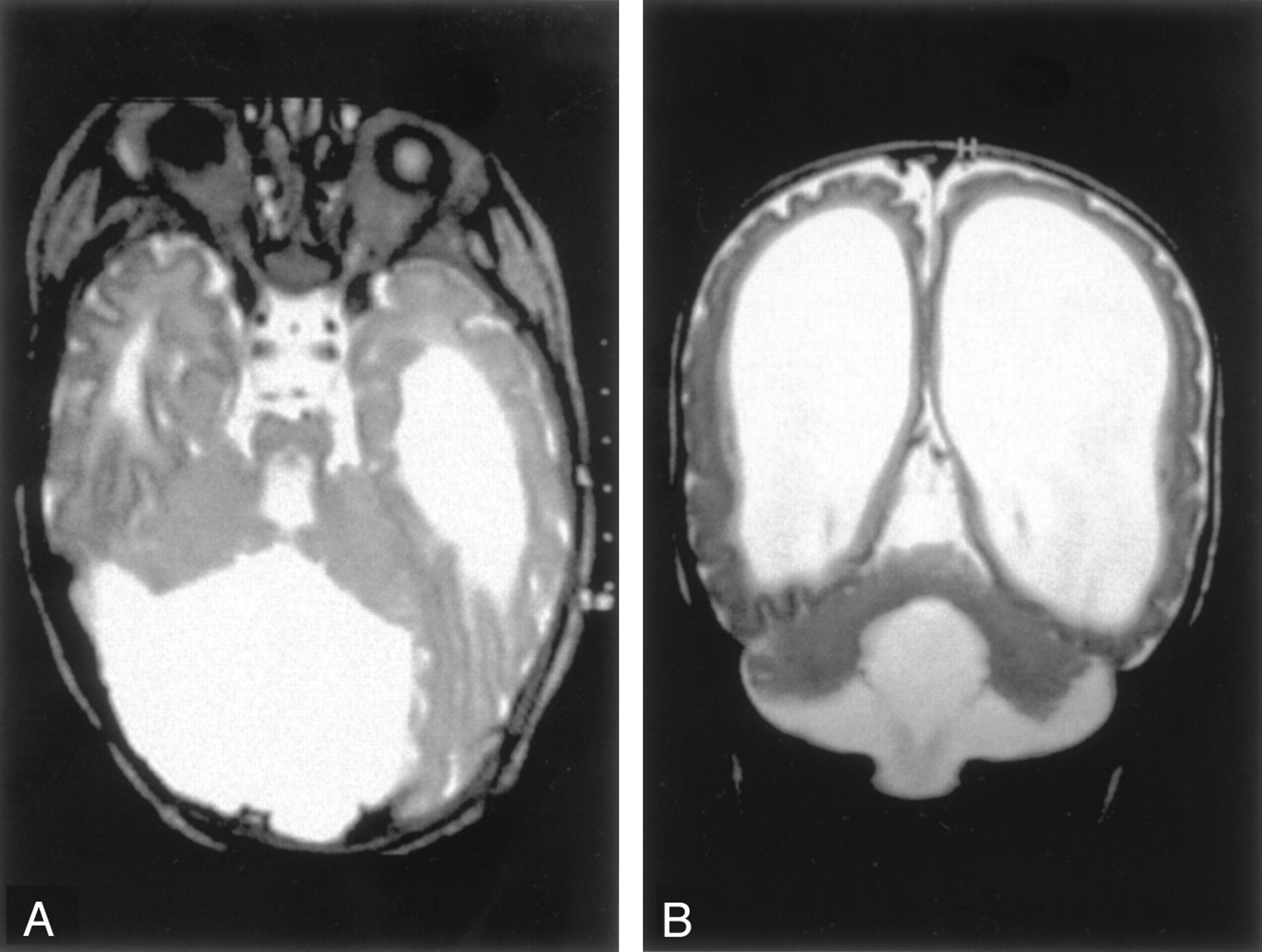
Congenital Muscular Dystrophy.—All four patients with Fukuyama congenital muscular dystrophy and one with muscle-eye-brain disease had cortical or subcortical cysts of the cerebellum, as have been described in the literature (28). Multiple small cortical folia, known in the literature as cerebellar polymicrogyria (28, 29) or cerebellar cortical dysplasia (30), were most pronounced through the superior cerebellum (Fig 6). In contrast, the cerebellum in both cases of Walker-Warburg syndrome and the other case of muscle-eye-brain disease appeared small and severely dysplastic (Fig 7). The patients with Walker-Warburg syndrome and with muscle-eye-brain disease were noted to have pontine hypoplasia with a midline longitudinal pontine cleft (Fig 7A). No vermis was identified in one of the patients with Walker-Warburg syndrome; however, the posterior fossa was not enlarged.
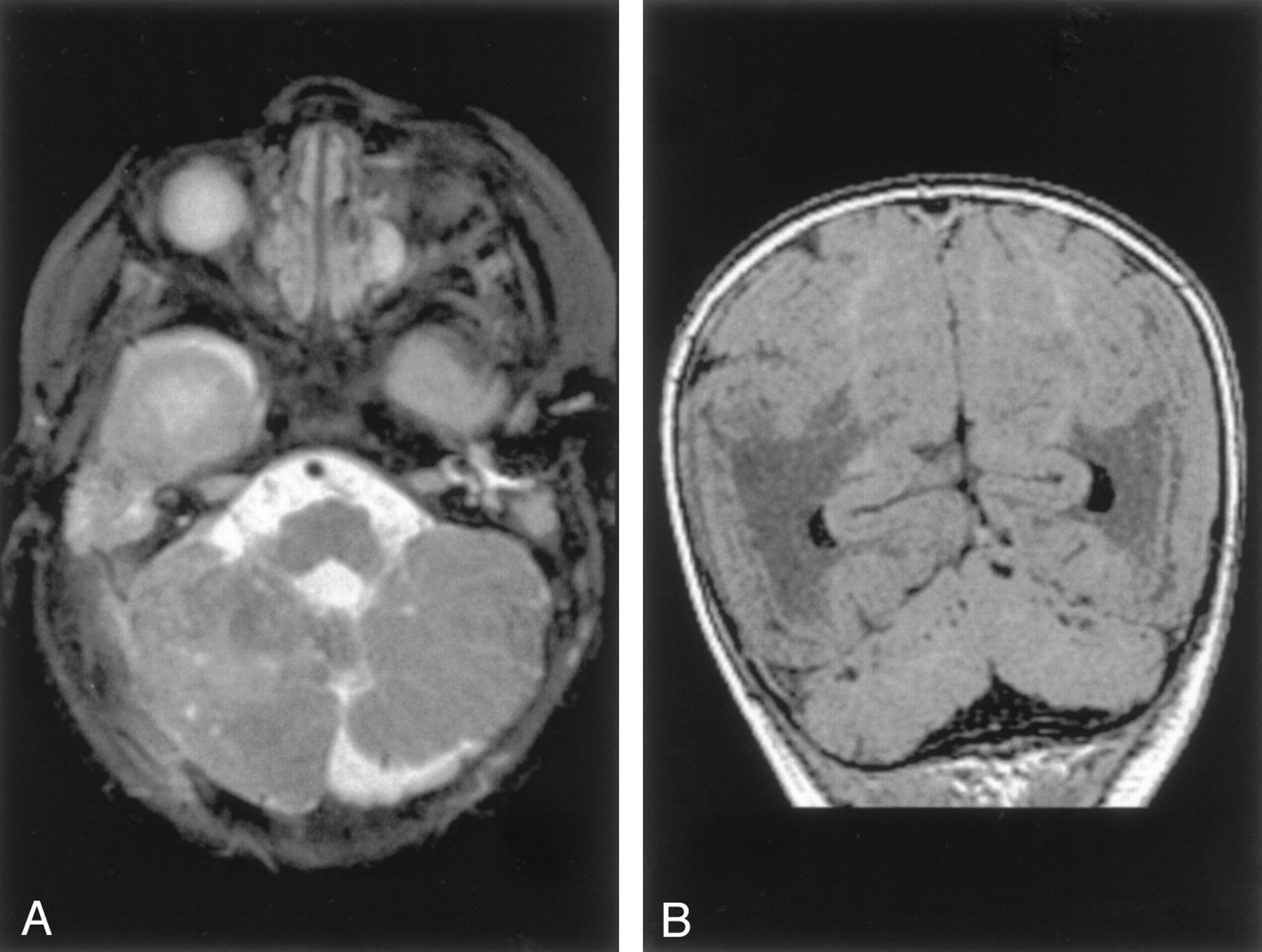
Cerebellar dysplasia in a 10-month-old patient with congential muscular dystrophy.
A, Axial spin-echo (3000/120) image shows abnormal folial pattern and small cysts in the cerebellar hemispheres.
B, Coronal gradient-echo (35/7) (theta of 45 degrees) image shows that the cysts are primarily in the superior aspects of the cerebellar hemispheres. Note the cobblestone cortex of the cerebrum.
Diffuse cerebellar dysplasia in a neonate with Walker-Warburg syndrome.
A, Axial spin-echo (3000/120) image shows small cerebellar hemispheres with dysplastic folial pattern.
B, Coronal spin-echo (600/16) image shows marked ventriculomegaly and cobblestone cortex.
Both patients with merosin-deficient muscular dystrophy displayed delayed myelination diffusely within the deep cerebral and cerebellar white matter, with mild inferior vermian hypogenesis but normal-appearing cortex in the cerebellar hemispheres. The patients with Fukuyama congenital muscular dystrophy and Walker-Warburg syndrome were noted to have cobblestone cortex, in some cases associated with polymicrogyria in the cerebral hemispheres (31, 32), whereas those with merosin-deficient congenital muscular dystrophy had diffuse supratentorial hypomyelination (33, 34).
Congenital Cytomegalovirus Infection.—The appearance of the cerebellar hemispheres in congenital cytomegalovirus infection varied directly with the appearance of the cerebral hemispheres. In the three patients with complete lissencephaly and thin cerebral cortex, the cerebella were small and shrunken, with thin cortex and shallow sulci. The hemispheres were extremely small, and the vermis was not seen at all; instead, a thin rim of tissue connected the dysplastic hemispheres (Fig 8). Cerebellar cortical calcification was present in one patient. The brain stems were small in approximate proportion to the respective cerebra and cerebella. In the three patients with diffuse polymicrogyria, the cerebella were nearly normal in size, with much milder dysplasia of the folial pattern. (For completeness, although not part of this study, those patients with only white matter injury had normal-appearing cerebella. Also for completeness, no unexpected cerebral anomalies were identified.)
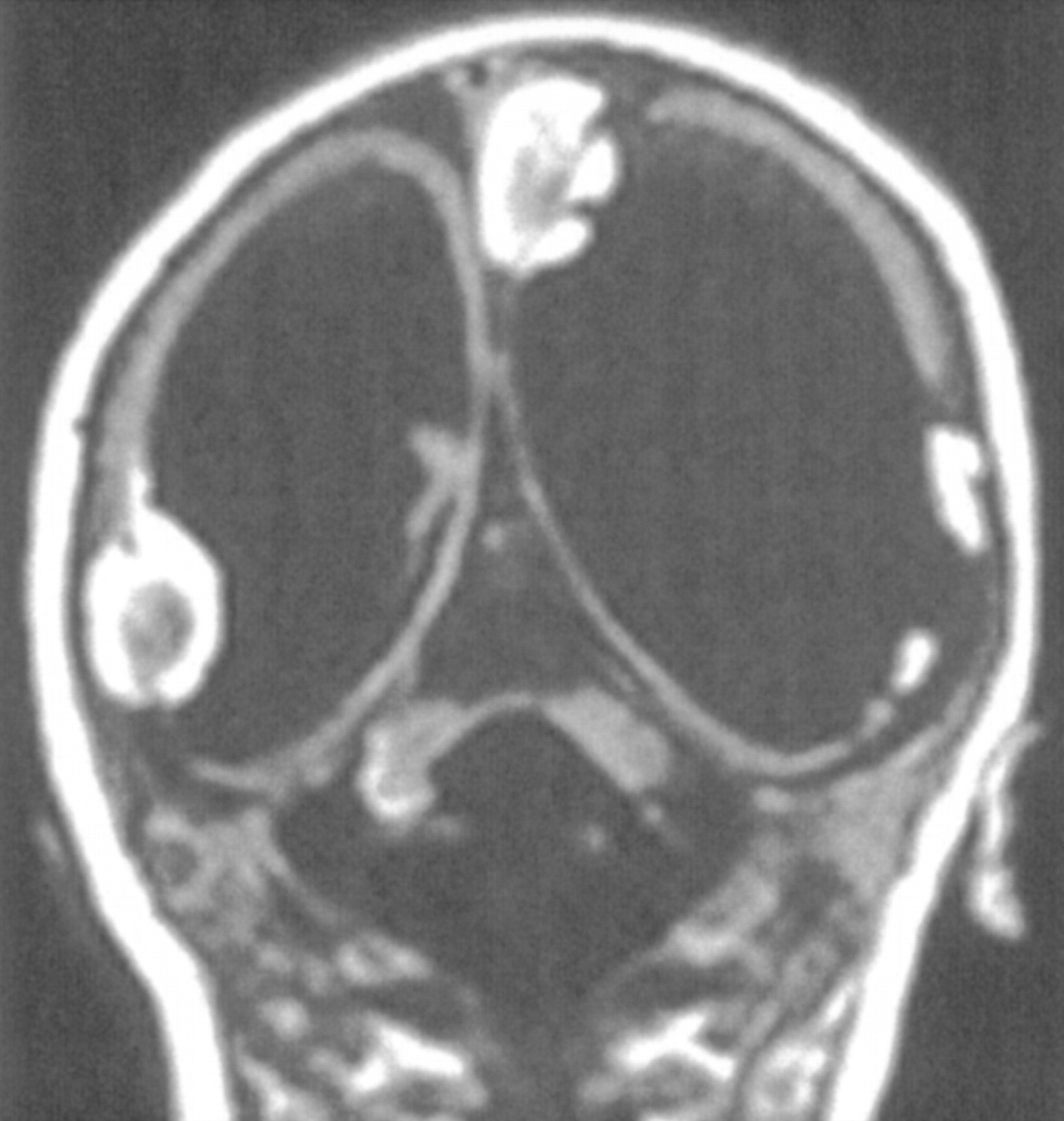
Cerebellar dysplasia in a neonate with congenital cytomegalovirus infection. Coronal spin-echo (600/16) image shows markedly small cerebellar hemispheres with almost no foliation. The hyperintensity of the right hemisphere was the result of calcification. Note the lissencephaly of the cerebrum with large calcifications.
Cerebellar Heterotopia.—One patient had gray matter heterotopia bilaterally in the corpus medullare of the cerebellum, associated with abnormal cortical foliation throughout both cerebellar hemispheres (Fig 9). In addition, the patient had fused colliculi and supratentorial anomalies, with reduced white matter volume in the right cerebral hemisphere and left occipital subcortical heterotopia and overlying polymicrogyria. The corpus callosum was normal.

Cerebellar cortical dysplasia with subcortical heterotopia in a 3-month-old patient.
A, Axial spin-echo (3000/120) image shows abnormal cerebellar cortical folial pattern and the presence of nodules of gray matter intensity (arrows) in the cerebellar white matter.
B, Coronal spin-echo (600/16) image shows that the nodule in the cerebellar white matter (arrows) remains isointense to gray matter on this T1-weighted image. The abnormal cerebellar folial pattern is again seen. In addition, the abnormality of the left cerebral hemisphere, which appears to represent polymicrogyria, is seen on this image.
Diffuse Dysplasia of the Cerebellar Cortex.—In one case, the entire cerebellum was dysmorphic, with bizarre, thick folia that were oriented vertically instead of the normal horizontal orientation (12). The brain stem was grossly normal. The supratentorial structures showed normal cortex and midline structures. Some subjective reduction in the volume of cerebral white matter was noted, along with some CSF-intensity cysts in the peritrigonal white matter.
Focal Dysplasias
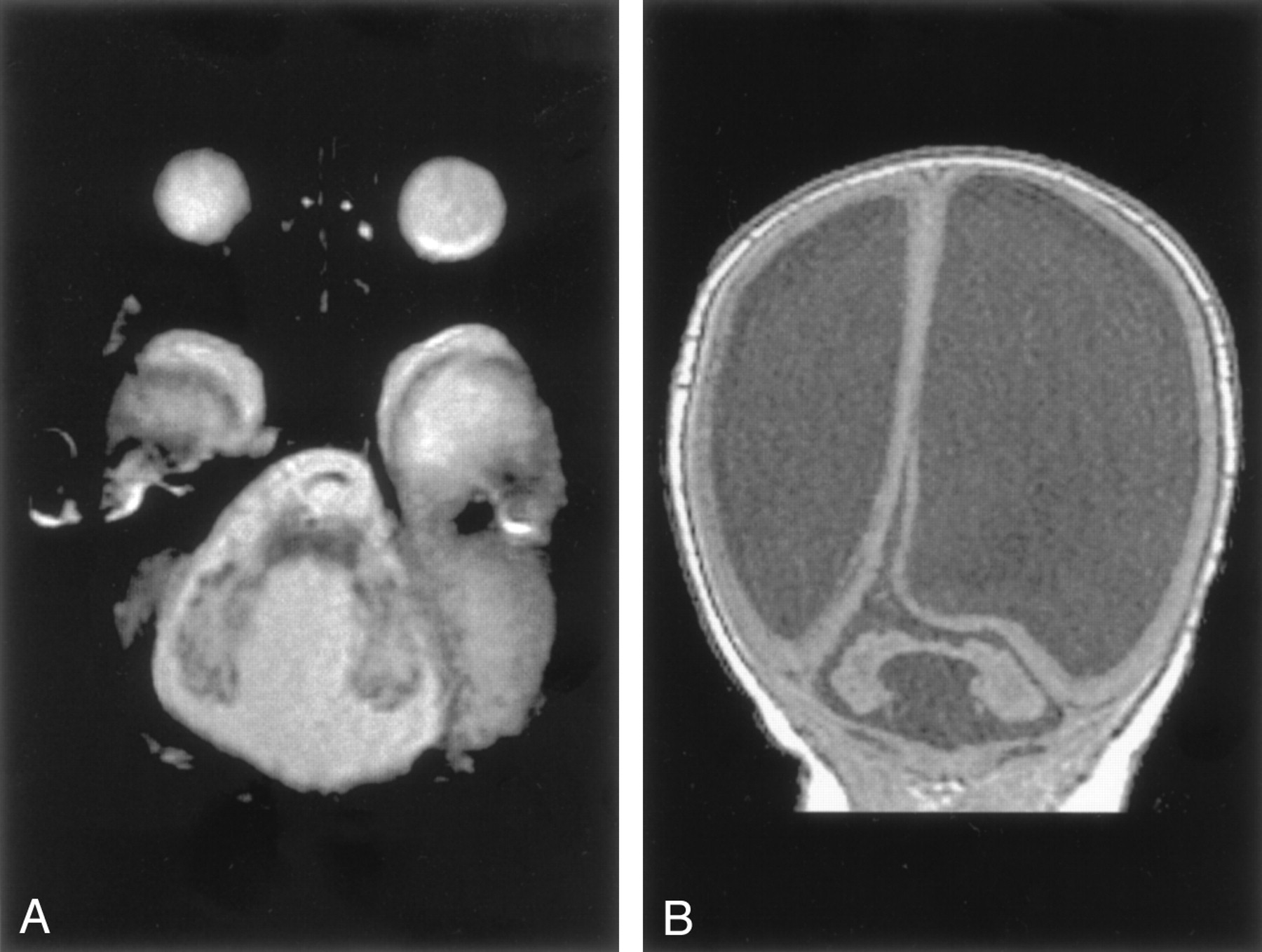
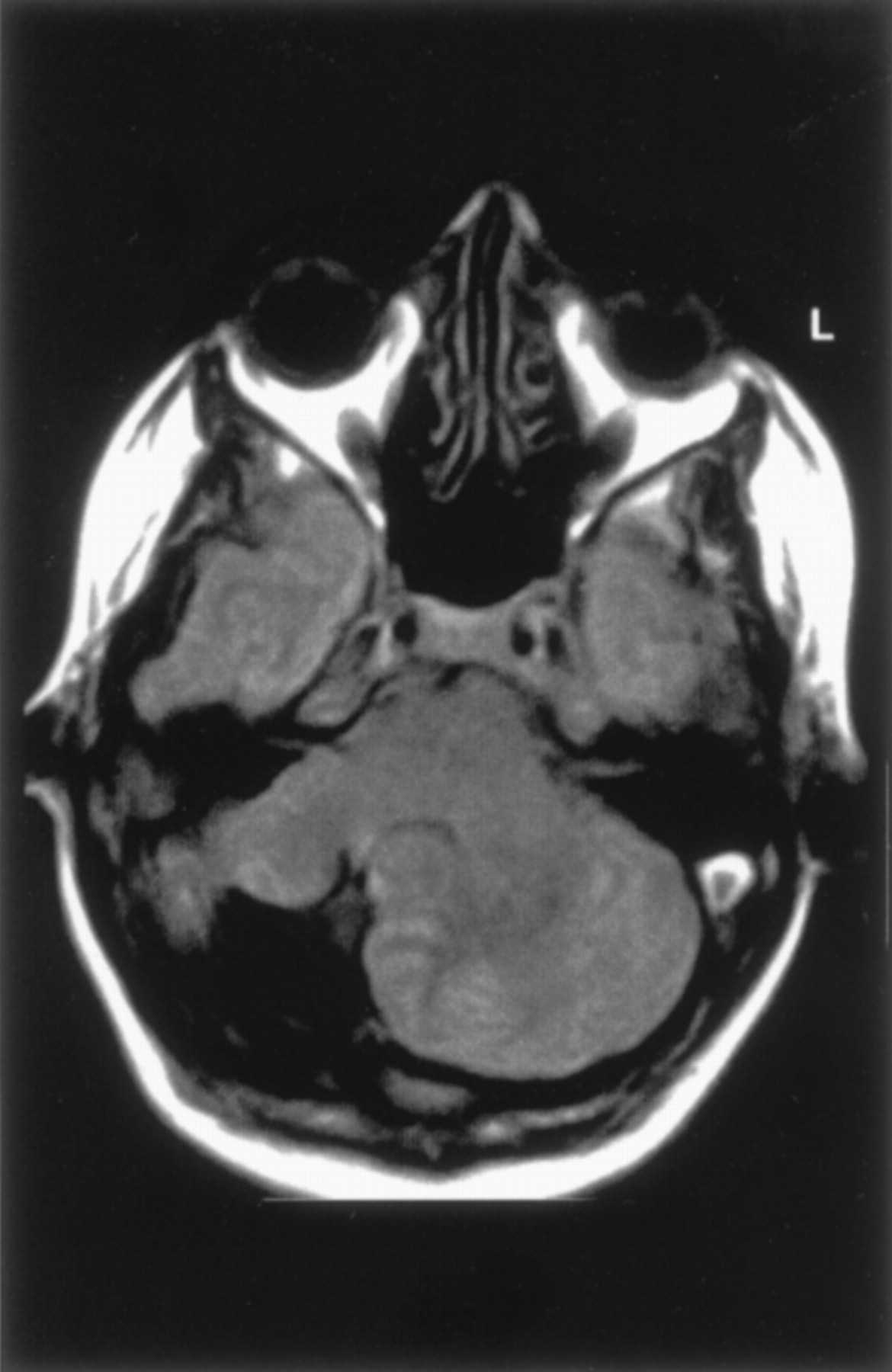
Rhombencephalosynapsis.—By definition, all patients with rhombencephalosynapsis had absence of the cerebellar vermis with midline fusion of the two cerebellar hemispheres (Fig 10) (11, 20). The dentate nuclei appeared fused in six and apposed, or perhaps partially fused, in the remaining two. The superior cerebellar peduncles were small, possibly hypoplastic. The degree of fusion between the superior and middle peduncles was difficult to assess in the absence of a high-resolution volumetric sequence. The superior colliculi were fused in two patients. In seven patients, overall cerebellar volume was normal or slightly reduced, and in the remaining patient, moderate cerebellar hypoplasia was present. The cerebral hemispheres showed subjectively reduced white matter volume in six patients, ventricular enlargement in five, and absence of the septum pellucidum in five. One patient had an infolding of dysplastic cerebral cortex, most likely polymicrogyria. The volume of the brain stem was also subjectively reduced in five patients; it could not be determined whether this reduction was the result of cerebellar anomalies, associated cerebral anomalies, or both.

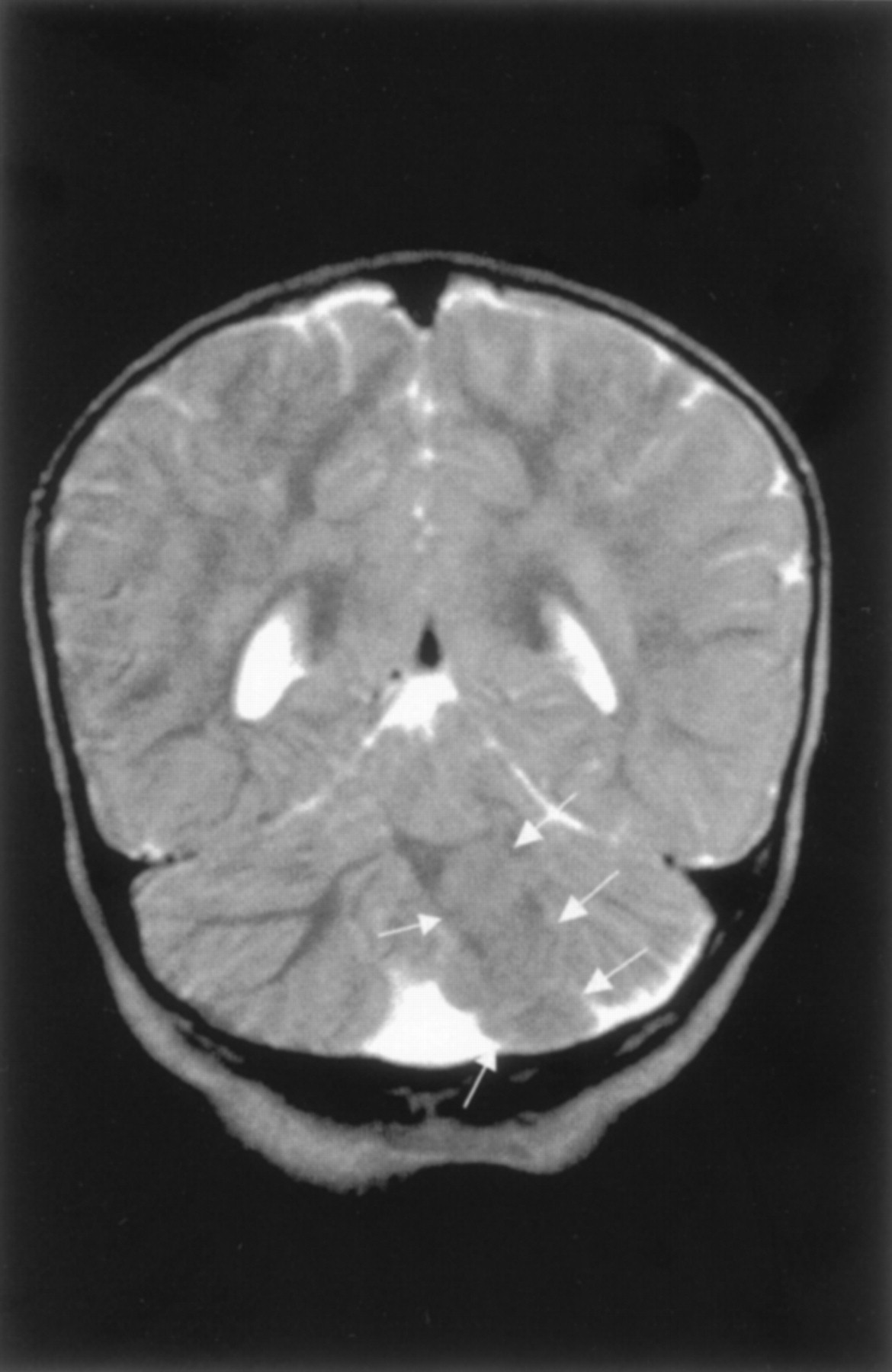
Rhombencephalosynapsis in a 6-year-old boy.
A, Sagittal spin-echo (550/15) image shows abnormal cerebellar vermis.
B, Axial spin-echo (2500/80) image shows continuity of the cerebral hemispheres across the midline without a midline cerebellar vermis. The cerebral hemispheres are abnormal, with reduced white matter and inward folding of the cortex as a result of ventriculoperitoneal shunting.
Molar Tooth Malformations.—Of the 12 patients with molar tooth malformations, good medical histories were available for only three; therefore, further classification according to the classification of Satran et al (5) was not possible. Nine of the patients had severe vermian hypoplasia. The rudimentary vermis was invariably dysplastic, with a midline cleft superiorly (Fig 11). In three patients, complete vermian agenesis appeared to be present. On axial images, the fourth ventricle had the typical “bat wing” or triangular configuration and communicated with the cisterna magna in each case. The flocculus appeared prominent in six cases. The cerebellar hemispheres appeared somewhat large, and the folial pattern seemed more vertical than normal. Whether this was the effect of the vermian anomaly on hemispheric orientation or whether the hemispheres were primarily dysplastic was uncertain.

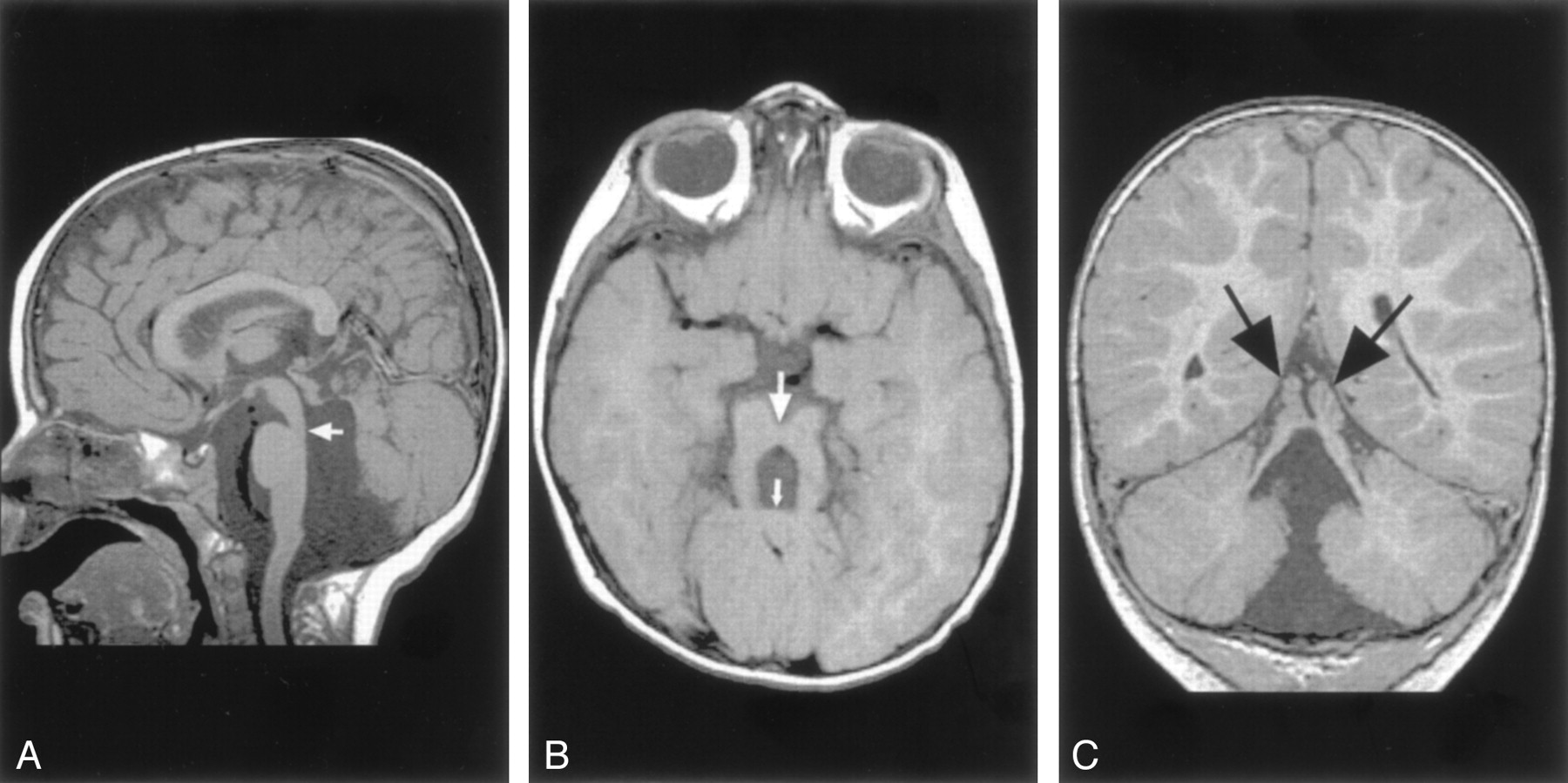
Molar tooth malformation in a 16-month-old patient with Joubert syndrome.
A, Sagittal spin-echo (550/11) image shows a small, dysplastic cerebellar vermis. The folial pattern is abnormal. Note the narrow isthmus (junction of the mesencephalon and pons) (arrow).
B, Axial spin-echo (550/15) image shows the very small vermis (small arrow), the broad, horizontal superior cerebellar peduncles, and the narrow isthmus (large arrow).
C, Coronal spin-echo (600/11) image shows a midline cleft separating the two dysplastic areas of vermis (arrows).
Several abnormalities were present at midbrain level. All patients had a deep interpeduncular fossa, thin isthmic region (pontomesencephalic junction) (Fig 11A and B), and thickened superior cerebellar peduncles that were perpendicular to the pons. These changes produced the so-called molar tooth appearance on axial view images (19) (Fig 11B). Variations in volume and shape at the cervicomedullary region were noted in eight patients. A reduction in pontine volume existed in two cases. The cerebral hemispheres appeared normal other than prominence of CSF spaces in three patients.
Lhermitte-Duclos-Cowden Gangliocytoma.—The images obtained in one patient showed the classic finding of a sharply defined, nonenhancing focal cerebellar mass in the left cerebellar hemisphere that showed prolonged T1 and T2 relaxation times, along with curvilinear stripes that were isointense to the cerebellar cortex. The mass resulted in moderate mass effect on the brain stem. Surgical exploration revealed thickened folia without obvious mass. Histologic examination of a biopsy specimen showed an outer layer of radial and superficial parallel myelinated fibers with an inner layer of dysplastic appearing neurons. Considering that the patient had hamartomata of the face and multiple other organs, and thus, had the diagnosis of Cowden syndrome, a confident diagnosis of dysplastic gangliocytoma of the cerebellum was made. The cerebral hemispheres were otherwise normal.
Other Focal Dysplasias.—One case of unilateral focal cerebellar hemispheric dysplasia was noted as an incidental finding in a 12-year-old boy who had mild head trauma. Another case of focal dysplasia involving a hemisphere and the vermis was observed in a 10-month-old male patient presenting with delayed motor development. In both of these patients, the lesions appeared as focal areas of disorganized architecture, one limited to the medial hemisphere (Fig 12) and the other involving the medial hemisphere and the adjacent vermis. The folial pattern of the affected regions was abnormal. The region of dysplasia had similar cortical signal intensity to the remainder of the cerebellar cortex, but the signal intensity of the dysplastic white matter was slightly altered (iso- or mildly hyperintense to adjacent normal cerebellar cortex and hyperintense to the remaining cerebellar white matter on T2-weighted images). The volume of the cerebellum was focally mildly reduced in the region of the dysplasia. The cerebral hemispheres were normal.

Focal cerebellar dysplasia in a 20-month-old female patient. Coronal view fast spin-echo (3500/112) image shows a focal region of abnormal foliation (arrows) in the inferomedial portion of the left cerebellar hemisphere.
Discussion
Cerebellar Embryology
As with all malformations, understanding of cerebellar malformations is greatly aided by an understanding of cerebellar development (Fig 13). In addition, classification is often facilitated by an understanding of embryology. Moreover, if genetics, embryology, and anatomy are all well understood, anatomic classification of malformations may eventually aid in determining molecular genetics.
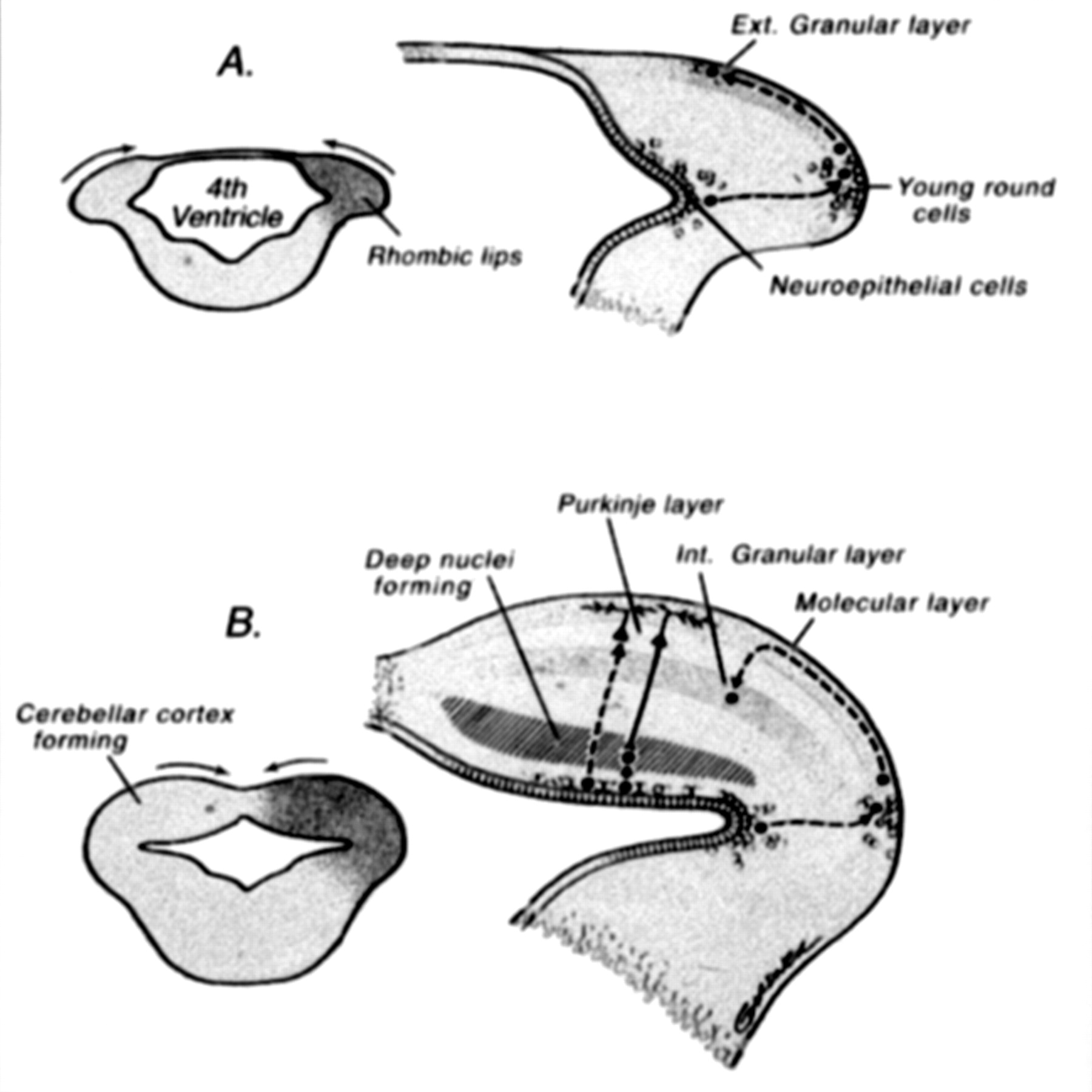
Drawings illustrate normal cerebellar development. During the 5th gestational week, cellular proliferation within the alar plates in conjunction with formation of the pontine flexure forms the rhombic lips. The neuroepithelial zones, in the roof of the fourth ventricle and the rhombic lips, are the locations of the germinal matrices where the cells of the cerebellum and many brain stem nuclei will form. Between 9 and 13 postconceptional weeks, the Purkinje cells of the cerebellar cortex and the neurons of the deep cerebellar nuclei migrate radially outward from this germinal matrix. In contrast, the neurons of the granular layer of the cerebellar cortex migrate tangentially from the germinal zone of the rhombic lips, over the cerebellar surface to form a transient external (Ext.) granular layer, which acts as a secondary germinal matrix. The external granular layer forms between the 10th and 11th postconceptional weeks and persists until approximately 15 months postnatal. The cells in the external granular layer proliferate, and the granule cell neuroblasts begin to migrate inward between clusters of homophilic Purkinje cells with the presumed aid of radial glial (Bergman) fibers, forming the internal (Int.) granular layer (reprinted with permission from Lippincott Williams & Wilkins [27]).
Early specification of cerebellar territory depends on the temporal and spatial expression of homeotic genes. A potent embryonic organizer within the neuroepithelium controls the early patterning of the prospective midbrain and anterior hindbrain. This is localized to a region of the isthmic constriction at the midbrain-hindbrain boundary and has been termed the m/h or isthmic organizer (35–37). Ongoing research about the genes and signaling molecules associated with the isthmic organizer exists. For instance, murine genes such as En1 and En2 seem to play a role in specifying cerebellar domain in the embryo (38). Targeted knockouts of these genes in mice produce cerebellar and collicular abnormalities (39–42). It is also clear that certain molecules that serve as cues for axon guidance in the developing brain, such as the netrins and their receptors (43) and the Wnt family of signaling molecules (44), play a role in cerebellar development by restricting the migration of cerebellar granule cells (45–47).
After the appearance of the pontine flexure at approximately the 5th gestational week, the fourth ventricle has a thin roof and laterally positioned dorsal (alar) plates (48, 49). During the 5th gestational week, cellular proliferation within the alar plates and formation of the pontine flexure form the rhombic lips. The neuroepithelial zones, in the roof of the fourth ventricle and the rhombic lips, are the locations of the germinal matrices where the cells of the cerebellum and many brain stem nuclei will form (45, 50). Between 9 and 13 postconceptional weeks, the Purkinje cells of the cerebellar cortex and the neurons of the deep cerebellar nuclei migrate radially outward from this germinal matrix. The identity and migration of the Purkinje cells depends on the expression of multiple genes, including Wnt3 (51). In contrast, the neurons of the granular layer of the cerebellar cortex migrate tangentially from the germinal zone of the rhombic lips, over the cerebellar surface, to form a transient external granular layer, which acts as a secondary germinal matrix (50, 52, 53). The external granular layer forms between the 10th and 11th postconceptional weeks and persists until approximately 15 postnatal months. The cells in the external granular layer proliferate, and the granule cell neuroblasts begin to migrate inward between clusters of homophilic Purkinje cells, with the presumed aid of radial glial (Bergman) fibers, forming the internal granular layer (48, 54, 55). Also formed at that time are the stellate and basket cells of the cerebellar cortex (45, 48, 49, 56, 57). Granule cell viability and migration are supported by several genes, the most important being Pax6, Zic1, and Math1, whereas the Bergman glia are preserved by the expression of Pax3 (58, 59).
Although multiple connections exist among cerebellar neurons, only two major afferent projections to the cerebellar cortex exist. Mossy fibers from the basis pontis and the spinal cord make contact and synapse with granule cells, whereas climbing fibers from the contralateral inferior olives make contact with Purkinje cell dendrites (45, 57, 60). In addition, axons from locus coeruleus, raphe nuclei, and substantia nigra synapse with Purkinje cells. The only efferent fibers from the cerebellar cortex are from the Purkinje cells, which connect with many brain stem nuclei, in addition to the cerebellar nuclei (61).
Classification Rationale
What is the importance of a classification of cerebellar anomalies? Some might (and indeed will) argue that a classification scheme does not necessarily aid diagnosis or prognostication. We disagree. For example, a classification such as the one proposed herein helps the clinician or imager who discovers a malformation for which he or she has no name. After determining (by defined criteria) whether the malformation represents hypoplasia or dysplasia and whether it is focal or generalized, it can be put into the proper category with similar malformations. By finding the outcome of similar malformations, the physician will have a better idea of the prognosis of the malformation that is being evaluated. More importantly, by grouping similar malformations according to a classification system, large numbers of identical malformations can be identified, allowing identification of mutations that cause malformations. This aids in genetic counseling, and by identifying the gene protein, in understanding how the brain is formed and which processes result in malformation.
The classifications that have been proposed for brain malformations largely ignore the status of the cerebellum, probably because cerebral anomalies are generally considered to be more important in determining cognitive and neurologic outcomes. However, this lack of attention to the cerebellum has resulted in a notable lag in our understanding of cerebellar anomalies as compared with our understanding of cerebral anomalies. Moreover, it is becoming increasingly clear that the cerebellum is important in cognition and motor function (62–66). Perhaps the ideal approach would be a classification system in which both supra- and infratentorial malformations are listed. All the patients with diffuse cerebellar dysplasia studied herein had anomalies of the cerebrum. However, in most of our patients with localized dysplasia or with hypoplasia/hypogenesis, the malformation was isolated to the posterior fossa, without associated supratentorial findings. Although our patients included three with lissencephaly and associated cerebellar hypoplasia, most cases of lissencephaly have normal cerebella (67). This relative paucity of combined cerebral-cerebellar malformations suggests that a meaningful classification combining infratentorial and supratentorial malformations does not seem practical at this stage. In the future, a molecular biologic classification, as suggested by Sarnat (68), based on the role(s) of the mutated gene(s) in the processes of brain development, may be the most practical way to classify brain anomalies, both cerebral and cerebellar. However, our knowledge of the genetic basis of malformations is still in an embryonic stage at present. Considering the current state of knowledge of these disorders, it seems most practical, at this time, to use a morphology-based classification. Separation of disorders by morphology has been used in the classification of cerebral disorders as well (69), and classification in this manner allows segregation of patients (particularly relatives) with like malformations, a process that can eventually lead to the identification of the causative mutations (70–72). It is hoped that the use of this classification system will allow similar progress to be made in understanding the genetic and molecular biologic basis of cerebellar development and maldevelopment.
Several methods could have been used as a basis for classification of cerebellar malformations. It seemed reasonable that the initial division separated dysplasia from hypoplasia, as the processes that result in these abnormalities are fundamentally different. We adopted the scheme proposed by Barkovich (27) to classify incompletely formed or small but otherwise normal-appearing cerebella as hypoplastic and cerebella with dysplastic folia and fissures as dysplastic. Hypoplastic cerebella can be considered to result from reduction or premature cessation of cell production or cell migration or of excessive apoptosis in the developing cerebellum. The reduction of cell production need not be generalized; it has been shown that depletion of Purkinje cells will cause a concomitant reduction in the granule cell population (73, 74). In contrast, dysplastic cerebella are considered to be the result of abnormal cell migration and cortical organization, with resultant distortions of folia and fissures. After the more fundamental differentiation of incomplete formation (hypoplasia) from abnormal formation (dysplasia), we incorporated topologic considerations to separate focal from generalized (diffuse) involvement. This separation was based on the assumption that focal processes are more likely to result from localized injury, whereas generalized involvement is more likely to result from genetic or metabolic causes. Another possibility was to separate median from lateral involvement, as suggested by Macchi and Bentivoglio (75); however, focal versus generalized classification was more practical. For example, some of the focal dysplasias in our study seemed to involve the medial hemisphere as well as the vermis; it was not clear whether this (clearly focal) condition should be classified as median or lateral. Thus, focal versus diffuse classification seemed to be a better solution. In addition, our separation of focal from generalized dysplasia led to the interesting observation that all patients with generalized cerebellar dysplasia had associated cerebral dysplasia, whereas patients with hypoplasia and focal dysplasia generally had normal cerebra.
In addition to the creation of a classification system, our analysis of cerebellar malformations added information concerning a number of cerebellar malformations. The Dandy-Walker malformation, for example, remains poorly understood, despite being extensively addressed in the literature. The disorder seems to be the result of a genetic predisposition, because the recurrence rate for siblings is 6% (76), and many associated malformations, such as agenesis of the corpus callosum, are known to be genetic. It remains to be determined which genes are mutated in this malformation and whether these genes are involved in the development of the cerebellum, the fourth ventricle, or both. The normal development of the fourth ventricle seems to be related to posterior fossa cyst formation. Bonnevie and Brodal (77) have shown that on the 11th fetal day in the normal mouse, the roof of the fourth ventricle is divided by a ridge of developing choroid plexus into anterior and posterior membranous areas. The theories proposed for the variable degree of enlargement of the fourth ventricle (and posterior fossa) focus on the severity of insult to the developing membranous area or to the extent of delay of opening of the foramen of Magendie (78, 79). Others have argued that because the anterior membranous area normally disappears before the fourth ventricular foramina open, maldevelopment of the cerebellar vermis is the most plausible explanation (80, 81). Other possibilities include persistence of Blake’s pouch, an extension of the fourth ventricle into the retrocerebellar subarachnoid space, which interferes with apposition of the cerebellar hemispheres inferiorly, and consequently, formation of the inferior vermis (2) and a combination of vermian and fourth ventricle developmental disorders (4).
Whether maldevelopment of the membranous area, vermis, or other as yet unknown region(s) is primarily responsible, a continuum of structural outcomes seems to exist. This continuum creates problems in attempting to classify individual cases. For example, when is the posterior fossa big enough to qualify for classification within the Dandy-Walker malformation continuum instead of vermian hypoplasia? In addition to the problems created by the combination of varying degrees of vermian hypoplasia and fourth ventricular enlargement, several well-documented cases of molar tooth malformations have characteristics of Dandy-Walker malformations (5); how are these best classified? Is the Dandy-Walker malformation merely the result of hydrocephalus superimposed on any degree of vermian hypoplasia or dysgenesis? If so, it may be a mixture of many different cerebellar malformations, the details of which have been blurred by the association with the fourth ventricular cyst. This mixture may explain why some Dandy-Walker malformations have only mild inferior vermian hypoplasia and others have diffuse cerebellar hypoplasia or cerebellar dysplasia; why some have pontine anomalies and others have normal appearances of their pons; and why some have multiple supratentorial anomalies and others have normal cerebra. The heterogeneity may explain why this malformation remains so difficult to define and classify 80 years after its description (82).
Pontocerebellar hypoplasia is a form of generalized cerebellar hypoplasia in the presence of a small volume pons and a characteristic clinical presentation. Barth et al (83, 84) divided this condition into two groups: type I with associated spinal anterior horn degeneration and type II with progressive microcephaly and extrapyramidal manifestations such as chorea and dystonia. The difficulty with using this nomenclature is that many diffuse cerebellar anomalies have associated pontine hypoplasia; we found small pons associated with both cerebellar hypoplasia and cerebellar dysplasia, and we found small pons in cerebellar malformations associated with diffuse cerebral anomalies and those associated with normal cerebra. This association is present possibly because the rhombic lips, the germinal zone from which newly generated cells migrate to the transient external granular layer of the cerebellar cortex, also serves as the germinal zone for the neurons that form the ventral pontine nuclei (44). Moreover, the external granular layer also functions as a germinal zone and produces (in addition to more cerebellar granule cells) cells that will populate the rostral hindbrain, including the locus coeruleus and pontine reticular formation (50). Thus, the association of pontine and cerebellar hypoplasia is common. As a result, although we agree that types I and II pontocerebellar hypoplasia, as recently defined (83, 84) are probably distinct entities, the names are confusing and unfortunate. We would recommend renaming these disorders more descriptively on the basis of the clinicoradiologic syndrome.
Dysplasia can be defined as deranged tissue development (85). Although we could not verify that all the patients we classified as having dysplastic cerebella had histologic abnormalities, all had abnormal folial patterns, ranging from absence of fissures to large folia with shallow fissures to foliation in abnormal directions. The two patients with lissencephaly and callosal agenesis both had very small cerebella that were difficult to evaluate (Fig 5). We classified them as dysplastic, because the fissures were shallow, and because many of the same processes and molecules that guide cerebral cortical development are at work in cerebellar development, as well (45). Thus, dysplasia seemed more likely than hypoplasia. This entity, lissencephaly with callosal agenesis and cerebellar dysplasia, appears to be still another form of classical lissencephaly, to be added to those associated with LIS1 (86), DCX (71, 87), and RELN (88) mutations; none of the latter are typically associated with either callosal agenesis or very small cerebella.
The congenital muscular dystrophies are well known to cause generalized dysplasia of the cerebellum (28, 29). It is interesting to note the cortical and retinal abnormalities in the congenital muscular dystrophies are thought to be related to over-migration of neurons through a defective glial limiting membrane (29, 32, 89). Furthermore, fukutin, the gene that is mutated in Fukuyama congenital dystrophy, is expressed in the Cajal-Retzius cells, components of the molecular layer (layer 1 of the cortex) that are theorized to have a role in establishing the glial limiting membrane (90, 91). Thus, it is reasonable to suspect that the over-migration and dysplasia of the Purkinje and granule cells in the subpial region of the cerebellar cortex in the congenital muscular dystrophy results from a defective glial limiting membrane of the cerebellum. Mechanisms of dysplasia in our other cases of diffuse dysplasia, those with congenital cytomegalovirus infection and those with unknown diagnoses, are unclear. In cytomegalovirus, the cause may be destructive. Of interest, patients with Walker-Warburg syndrome have been described as having Dandy-Walker malformation (92). One of our patients had absence of the cerebellar vermis, in addition to severe dysplasia of the cerebellum, but no enlargement of the posterior fossa was seen. This again brings up the question of whether the Dandy-Walker malformation continuum concept is useful or whether it succeeds more in obfuscating the details underlying the cerebellar malformations.
Those malformations classified as focal dysplasia were a diverse group, including rhombencephalosynapsis, the molar tooth malformations, and a number of unclassifiable cases with localized distortions of the cerebellar folial pattern. Distinctive morphologic features of rhombencephalosynapsis include lack of development of the normal midline structures, primarily in the posterior fossa (midline mesencephalic structures in addition to the cerebellar vermis), with resultant fusion of the cerebellar hemispheres and colliculi and apposition or fusion of the dentate nuclei. Supratentorial anomalies tend to dominate the clinical picture (11, 93, 94). The embryogenesis of this disorder is uncertain. During normal development, the cerebellar hemispheres appear initially to be separate. The apposition of the cerebellar hemispheres in the dorsal midline is thought to result in fusion, with local induction of the cells that form the vermis (95). The mechanisms by which the vermian cells differentiate are not known. Recent work strongly suggests that other disorders characterized by absence of midline structures, such as holoprosencephaly, result from a failure of induction of the structures that normally compose the midline rather than fusion of structures that are induced to form in the paramedian brain (96, 97). In view of the absence of the septum pellucidum and the absence of midline structures in the posterior fossa, the possibility exists that rhombencephalosynapsis is caused by malexpression of gene expression in early patterning centers of the brain. Along these lines, it is interesting to note that the FGF8 gene is active both in the rostrodorsal cerebral midline (in the region of the septum) and in the isthmic organizer (where the cerebellar anlage is induced) (98). Whatever the cause, rhombencephalosynapsis is a developmental disorder of the midline, with relative preservation of lateral structures and is best classified as a focal dysplasia.
The molar tooth malformations are named for the characteristic appearance of the midbrain on axial images of affected patients (19, 99). Patients with this mesencephalic anomaly have a distinctive hindbrain malformation that was first described by Joubert et al in 1969 (6). For a long time, all malformations with this appearance were called Joubert syndrome. Recently, it has become apparent that the clinical manifestations of the patients with this posterior fossa malformation are variable (5). Some patients have renal anomalies (juvenile nephronophthisis or multicystic dysplastic kidney), some have ocular anomalies (retinal dysplasias and colobomata), some have hepatic fibrosis and cysts; these malformations have previously been given names such as Arima syndrome, COACH syndrome, and Senior-Löken syndrome (5). Most of these patients have a well-defined clinical syndrome associated with the hindbrain malformation, characterized by episodic hyperpnea, abnormal eye movements, ataxia, and mental retardation (7, 100). A few authors (6, 101–103) have provided neuropathalogic accounts of this malformation. Of considerable interest is the finding of a lack of decussation of the superior cerebellar peduncles, central pontine tracts, and corticospinal tracts. This finding raises the possibility of the lack of decussation of these pathways being the primary anomaly and the associated dysplasia of the vermis, cerebellar nuclei, and brain stem nuclei being secondary. Whatever the cause, the vermis is clearly dysplastic (based on both pathologic and imaging criteria), and the dysplasia appears to be medial, and therefore, focal.
Finally, our series shows that cortical dysplasia and subcortical heterotopia can be identified in the cerebellum, as they can in the cerebrum. Imaging findings of these anomalies have not been previously described in the radiology literature to our knowledge, although they have been recognized and described in the pathology literature (30, 67). The pathologists found, as we did, that diffuse cerebellar dysplasias commonly accompany extensive cerebral lesions, such as congenital muscular dystrophies or diffuse cerebral polymicrogyria, but may be found as incidental lesions in otherwise normal brains (30). The pathology literature also provides some detail regarding the focal dysplastic lesions, although more information is available regarding dysplastic lesions that are associated with cerebral anomalies (such as rhombencephalosynapsis and the molar tooth malformation) than regarding isolated focal dysplasias. We do not have the histologic details of the cases of isolated focal cerebellar cortical dysplasias, but we observed focal disorganization of the cerebellar cortex and reduced volume of underlying white matter, findings similar to those observed in focal cortical dysplasia of the cerebral cortex (27, 104). The mechanism by which the dysplasia develops is not certain; possibilities include genetically or environmentally disturbed migration of the Purkinje or granule cells and or improper signaling of the cellular or interstitital signals that guide developing cells and axons (45). Similarly, it is not certain whether the subcortical heterotopias are the result of under-migration of Purkinje cells, over-migration of granule cells, or impairment of programmed cell death. Of note, cerebellar cortical dysplasia with subcortical cerebellar heterotopia has been discovered in mice with mutations of the Unc5h3 gene, which codes for a receptor for netrin 1, an important guidance molecule during brain development (46, 47). It seems likely that focal cerebellar dysplasias are more common than is generally recognized. Moreover, in view of the growing body of evidence suggesting that the cerebellum has a role in motor learning and cognition (62, 63, 65, 105) these malformations might, with a higher index of suspicion, be recognized more commonly on imaging studies of children with developmental disabilities.
Conclusion
A retrospective review of 70 MR imaging studies obtained in patients with cerebellar anomalies suggests that these malformations can be organized according to a rational scheme. In this article, we propose such a scheme. Although the classification will undoubtedly need revision as our experience and understanding increases, this and subsequent schemes may help to classify similar malformations, and ultimately, to aid in the identification of the genetic and molecular biologic processes underlying cerebellar formation and malformation.
Acknowledgments
The authors thank Dr. Wally Peck, Professor Danielle Baleriaux, Dr. R. Devasthali, Dr. Clark Carrol, Dr. Paolo Galluzzi, Dr. Chris Walsh, and Dr. David Seidenwurm, who kindly supplied the case material that was used in this study.
References
- Received September 6, 2001.
- Accepted after revision March 14, 2002.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Cerebellar Heterotopias: Expanding the Phenotype of Cerebellar Dysgenesis in CHARGE Syndrome
- Variable brain phenotype primarily affects the brainstem and cerebellum in patients with osteogenesis imperfecta caused by recessive WNT1 mutations
- Normal Fetal Posterior Fossa in MR Imaging: New Biometric Data and Possible Clinical Significance
- Long-term developmental outcome of children with a fetal diagnosis of isolated inferior vermian hypoplasia
- Inherited Cerebellar Ataxia in Childhood: A Pattern-Recognition Approach Using Brain MRI
- Pontocerebellar hypoplasia type 1: Clinical spectrum and relevance of EXOSC3 mutations
- GPR56-Regulated Granule Cell Adhesion Is Essential for Rostral Cerebellar Development
- Mid-brain molar tooth sign: expanding the clinical spectrum
- Recent advances in the molecular pathology, cell biology and genetics of ciliopathies
- Mid-brain molar tooth sign: expanding the clinical spectrum
- Predominant dystonia with marked cerebellar atrophy: A rare phenotype in familial dystonia
- Magnetic Resonance Imaging of the Kinked Fetal Brain Stem: A Sign of Severe Dysgenesis
- Imaging the child with developmental delay
- The magnetic resonance revolution in brain imaging: impact on neonatal intensive care
- Mutations in the oligophrenin-1 gene (OPHN1) cause X linked congenital cerebellar hypoplasia