
Abstract
Editorial expression of concern:
In the May 2021 edition, the American Journal of Neuroradilogy published the article “MRI-Based Deep-Learning Method for Determining Glioma MGMT Promoter Methylation Status” by Yogananda CGB, et al.1 On August 22, 2022, the authors self-reported data errors related to the computer code and the training and testing data sets. The authors are now in the process of re-evaluating the accuracies using the correct test set. This notice of concern is to inform readers about these possible issues related to this articles results. After additional tests from the authors on the correct dataset are available, we will determine what additional action is warranted, such as an erratum.
1. Yogananda CGB, Shah BR, Nalawade SS, et al. MRI-based deep-learning method for determining glioma MGMT promoter methylation status. American Journal of Neuroradilogy. 2021;42(5):845-852. doi:10.3174/AJNR.A7029
BACKGROUND AND PURPOSE: O6-Methylguanine-DNA methyltransferase (MGMT) promoter methylation confers an improved prognosis and treatment response in gliomas. We developed a deep learning network for determining MGMT promoter methylation status using T2 weighted Images (T2WI) only.
MATERIALS AND METHODS: Brain MR imaging and corresponding genomic information were obtained for 247 subjects from The Cancer Imaging Archive and The Cancer Genome Atlas. One hundred sixty-three subjects had a methylated MGMT promoter. A T2WI-only network (MGMT-net) was developed to determine MGMT promoter methylation status and simultaneous single-label tumor segmentation. The network was trained using 3D-dense-UNets. Three-fold cross-validation was performed to generalize the performance of the networks. Dice scores were computed to determine tumor-segmentation accuracy.
RESULTS: The MGMT-net demonstrated a mean cross-validation accuracy of 94.73% across the 3 folds (95.12%, 93.98%, and 95.12%, [SD, 0.66%]) in predicting MGMT methylation status with a sensitivity and specificity of 96.31% [SD, 0.04%] and 91.66% [SD, 2.06%], respectively, and a mean area under the curve of 0.93 [SD, 0.01]. The whole tumor-segmentation mean Dice score was 0.82 [SD, 0.008].
CONCLUSIONS: We demonstrate high classification accuracy in predicting MGMT promoter methylation status using only T2WI. Our network surpasses the sensitivity, specificity, and accuracy of histologic and molecular methods. This result represents an important milestone toward using MR imaging to predict prognosis and treatment response.
ABBREVIATIONS:
- IDH
- isocitrate dehydrogenase
- MGMT
- O6-methylguanine-DNA methyltransferase
- PCR
- polymerase chain reaction
- T2WI
- T2 weighted Images
- TCGA
- The Cancer Genome Atlas
- TCIA
- The Cancer Imaging Archive
O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation is a molecular biomarker of gliomas that has prognostic and therapeutic implications. Unlike isocitrate dehydrogenase (IDH) mutations and 1p/19q co-deletions, MGMT promoter methylation is an epigenetic event. Epigenetic events are functionally relevant but do not involve a change in the nucleotide sequence. Therefore, while MGMT promoter methylation is an important prognostic marker, it does not define a distinct subset of gliomas. MGMT is a DNA repair enzyme that protects normal and glioma cells from alkylating chemotherapeutic agents. The methylation of the MGMT promoter is an example of epigenetic silencing, which results in a loss of function of the MGMT enzyme and its protective effect on glioma cells. The survival benefit incurred by MGMT promoter methylation in patients treated with temozolomide (TMZ) was determined in 2005.1 Subsequent work by Stupp et al2 has shown that in patients who received both radiation and temozolomide, MGMT promoter methylation improved median survival compared with patients with unmethylated gliomas (21.7 versus 12.7 months).2 Long-term follow-up from that initial study has further substantiated the survival benefit.2,3 As a result, determining MGMT promoter methylation status is an important step in predicting survival and determining treatment.
Currently, the only reliable way to determine MGMT promoter methylation status requires analysis of glioma tissue obtained either via an invasive brain biopsy or following open surgical resection. Surgical procedures carry the risk of neurologic injury and complications. Therefore, considerable attention has been dedicated to developing noninvasive, image-based diagnostic methods to determine MGMT promoter methylation status. A meta-analysis of MR imaging features demonstrated that glioblastomas with methylated MGMT promoters were associated with less edema, high ADC, and low perfusion. However, the summary sensitivity and specificity of these clinical features was only 79% and 78%, respectively.4 Although multiple radiomic approaches have also been attempted for MGMT prediction, none, to date, have achieved accuracies sufficient for clinical viability.5⇓⇓⇓-9 Sasaki et al10 attempted to establish an MR imaging–based radiomic model for predicting MGMT promoter status of the tumor, but it reached a predictive accuracy of only 67%. Wei et al11 extracted radiomic features from the tumor and peritumoral edema using multisequence, postcontrast MR imaging but only achieved an accuracy of 51%–74% in predicting MGMT promoter methylation status in astrocytomas. Drabycz et al5 performed texture analysis on MR images to predict MGMT promoter methylation status in glioblastoma, but it reached an accuracy of only 71%. Korfiatis et al9 combined texture features with supervised classification schemes as potential imaging biomarkers for predicting the MGMT methylation status of glioblastoma multiforme, but they achieved a sensitivity and specificity of only 0.803 and 0.813, respectively. Ahn et al7 used dynamic contrast-enhanced MR imaging and diffusion tensor imaging to predict MGMT promoter methylation in glioblastoma, but this method achieved a sensitivity and specificity of only 56.3% and 85.2%, respectively.
Recent advances in deep learning methods have also been used for noninvasive, image-based molecular profiling. Our group has previously demonstrated highly accurate, MR imaging–based, voxelwise, deep learning networks for determining IDH classification and 1p/19q co-deletion status using only T2WI.12,13 The benefits of using T2WIs are that they are routinely acquired, they can be obtained quickly, and high quality T2WI can even be obtained in the setting of motion degradation. Because MGMT promoter methylation in gliomas is such an important biomarker, we sought to develop a highly accurate, fully automated deep learning 3D network for MGMT promoter determination of methylation status using only T2WI.
MATERIALS AND METHODS
Data and Preprocessing
Multiparametric MR images of patients with brain gliomas were obtained from The Cancer Imaging Archive (TCIA) data base.14,15 The genomic information was obtained from both The Cancer Genome Atlas (TCGA) and TCIA data bases.14,16,17 Subject datasets were screened for the availability of preoperative MR images, T2WI, and known MGMT promoter status. The final dataset of 247 subjects included 163 methylated cases and 84 unmethylated cases. TCGA subject identification, MGMT status, and tumor grade are listed in Table 1 of the Online Supplemental Data.
Cross-validation results
Tumor masks for 179 subjects were available through previous expert segmentation.18⇓-20 Tumor masks for the remaining 68 subjects were generated by our previously trained 3D-IDH network and were reviewed by 2 neuroradiologists for accuracy.20 These tumor masks were used as ground truth for tumor segmentation in the training step. Ground truth whole-tumor masks for methylated and unmethylated MGMT promoter type were labeled with 1's and 2's, respectively (Fig 1). Data preprocessing steps included the following: 1) the Advanced Normalization Tools software package (http://stnava.github.io/ANTs/) affine coregistration21 to the SRI24 T2 template,22 2) skull stripping using the Brain Extraction Tool (BET; http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/BET)23 from FSL,23⇓⇓-26 3) removing radiofrequency inhomogeneity using N4 Bias Field Correction (https://simpleitk.readthedocs.io/en/master/link_N4BiasFieldCorrection_docs.html),27 and 4) normalizing intensity to zero-mean and unit variance. The preprocessing took <5 minutes per dataset.
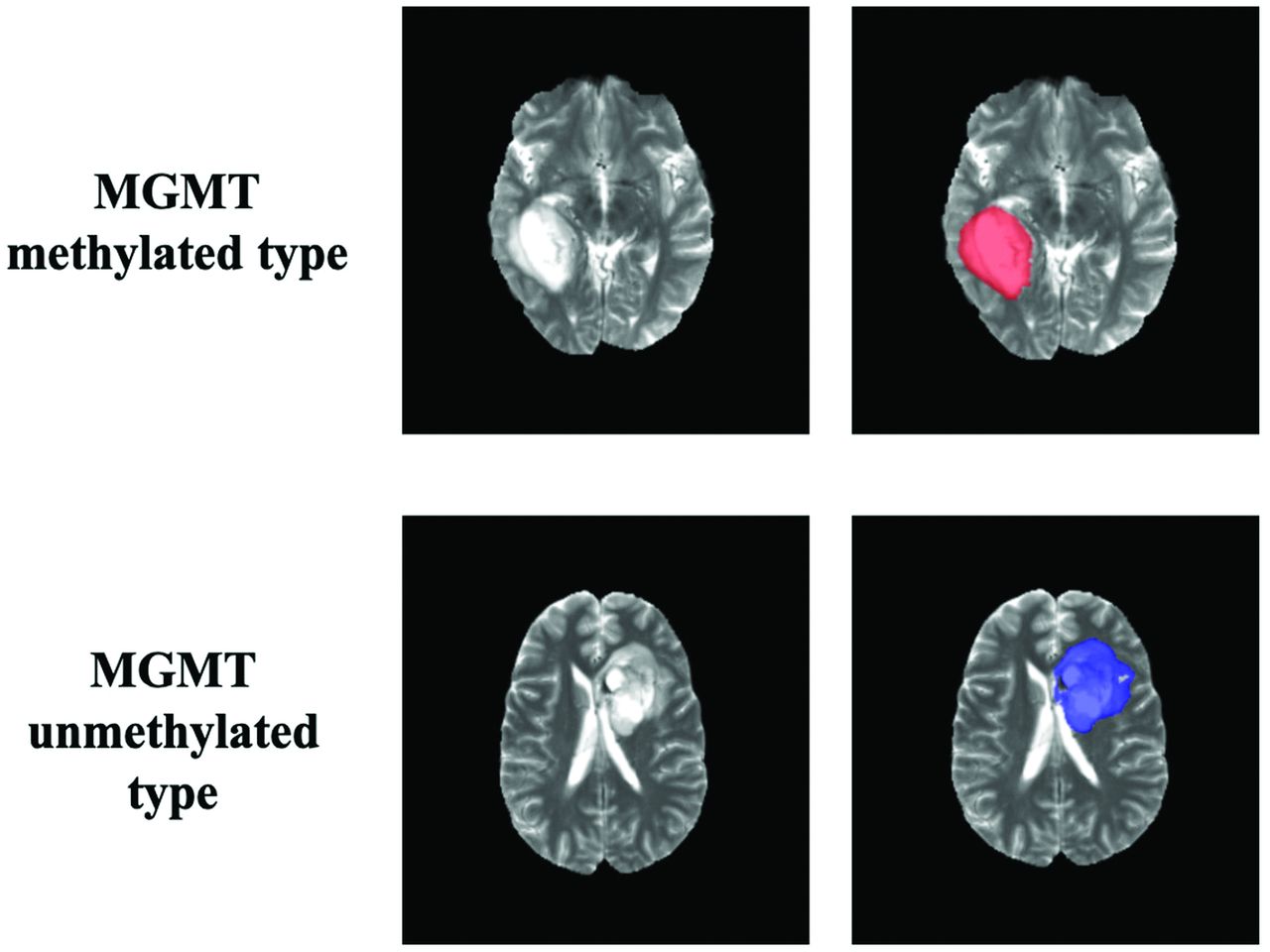
Ground truth whole-tumor masks. Red voxels represent methylated MGMT promoter status (values of 1) and blue voxels represent unmethylated MGMT promoter status (values of 2). The ground truth labels have the same MGMT promoter status for all voxels in each tumor.
Network Details
Transfer learning for determination of MGMT promoter status was implemented using our previously trained 3D-IDH network.20 The decoder part of the network was fine-tuned for a voxelwise dual-class segmentation of the whole tumor, with 1 and 2 representing methylated and unmethylated MGMT promoter types, respectively. The network architecture is shown in Fig 2B. A detailed schematic of the network is provided in the Online Supplemental Data.

A, MGMT-net overview. Voxelwise classification of MGMT promoter status is performed to create 2 volumes (methylated and unmethylated MGMT promoter). Volumes are combined using Dual Volume Fusion to eliminate false-positives and generate a tumor-segmentation volume. Majority voting across voxels is used to determine the overall MGMT promoter status. B, Network architecture for MGMT-net. 3D-dense-UNets are used with 7 dense blocks, 3 transition-down (TD) blocks, and 3 transition-up (TU) blocks. Conv indicates convolution layer.
Network Implementation and Cross-Validation
To generalize the network's performance, we performed a 3-fold cross-validation. The dataset of 247 subjects was randomly shuffled and distributed into 3 groups (approximately 82 subjects for each group). Group 1 had 82 subjects (54 methylated, 28 unmethylated), group 2 had 83 subjects (55 methylated, 28 unmethylated), and group 3 had 82 subjects (54 methylated, 28 unmethylated). The 3 groups alternated among training, in-training validation, and held-out testing groups so that each fold of the cross-validation was a new training phase based on a unique combination of the 3 groups. The network uses the in-training validation dataset to evaluate its learning after each training round and updates model parameters to improve performance. However, the network performance is reported only on the held-out testing group for each fold because it is never seen by the network. The group membership for each cross-validation fold is listed in the Online Supplemental Data.
Seventy-five percent overlapping 3D patches (size: 32 × 32 × 32 voxels) were extracted from the training and in-training validation dataset. The patch extraction was performed as a translation in the x-y-z-plane. During training, only patches with at least 1 tumor voxel were included; thus, the number of patches included per training cases varied depending on the size of the tumor. For testing however, the entire image was sampled, including background masked voxels (of value zero). No patch from the same subject was mixed with the training, in-training validation, or testing datasets to prevent the problem of data leakage.28,29 Data augmentation steps included horizontal and vertical flipping, random and translational rotation, the addition of salt and pepper noise, the addition of Gaussian noise, and projective transformation. Additional data augmentation steps included down-sampling images by 50% and 25% (reducing the voxel resolution to 2 and 4 mm3). The data augmentation provided a total of approximately 300,000 patches for training and 300,000 patches for in-training validation for each fold. The networks were implemented using the Tensorflow30 backend engine, the Keras31 Python package, and an Adaptive Moment Estimation optimizer (Adam).32 The initial learning rate was set to 10−5 with a batch size of 15 and maximal epochs of 100 for each fold.
MGMT-net outputs 2 segmentation volumes (V1 and V2), which are combined to generate the voxelwise prediction of methylated and unmethylated MGMT promoter tumor voxels, respectively. The 2 volumes are fused, and the largest connected component (the 3D-connected component algorithm in Matlab [MathWorks]) is obtained as the single tumor-segmentation map. Majority voting over the voxelwise classes of methylated or unmethylated type provided a single MGMT promoter classification for each subject. Tesla V100s, P100, P40, and K80 NVIDIA-GPUs were used to implement the networks. This MGMT promoter determination process is fully automated, and a tumor segmentation map is a natural output of the voxelwise classification approach.
Statistical Analysis
Statistical analysis of the network's performance was performed in Matlab and R statistical and computing software (http://www.r-project.org/). Network accuracies were evaluated using majority voting (ie, a voxelwise cutoff of 50%). The accuracy, sensitivity, specificity, positive predictive value, and negative predictive value of the model for each fold of the cross-validation procedure were calculated using this threshold. Receiver operating characteristic curves for each fold were generated separately. Dice scores were calculated to evaluate the tumor-segmentation performance of the networks. The Dice score calculates the spatial overlap between the ground truth segmentation and the network segmentation.
RESULTS
The network achieved a mean cross-validation testing accuracy of 94.73% across the 3 folds (95.12%, 93.98%, and 95.12% [SD, 0.66%]). Mean cross-validation sensitivity, specificity, positive predictive value, negative predictive value, and area under the curve for the MGMT-net was 96.31% [SD, 0.04%], 91.66% [SD, 2.06%], 95.74% [SD, 0.95%], 92.76% [SD, 0.15%], and 0.93 [SD, 0.03], respectively. The mean cross-validation Dice score for tumor segmentation was 0.82 [SD, 0.008] (Table). The network misclassified 4 cases for fold one, 5 cases for fold 2, and 4 cases for fold three (13 total of 247 subjects). Six subjects were misclassified as unmethylated, and 7, as methylated.
Receiver Operating Characteristic Analysis
The receiver operating characteristic curves for each cross-validation fold for the network are provided in Fig 3. The network demonstrated very good performance with high sensitivities and specificities.

Receiver operating characteristic (ROC) analysis for MGMT-net. Separate curves are plotted for each cross-validation fold along with corresponding area under the curve (AUC) value.
Voxelwise Classification
The network is a voxelwise classifier with the tumor segmentation map being a natural output. Figure 4 shows examples of the voxelwise classification for methylated and unmethylated MGMT promoter types, respectively. The volume-fusion procedure was effective in removing false-positives and improving the Dice scores by approximately 6%. We also computed the voxelwise accuracy for the network. The mean voxelwise accuracies were 81.68% [SD, 0.02%] for methylated type and 70.83% [SD, 0.04%] for unmethylated type.

A, An example of voxelwise segmentation for a tumor with a methylated MGMT promoter: native T2WI (a), ground truth segmentation (b), and network output after Dual Volume Fusion (c). Red voxels correspond to MGMT methylated class, and blue voxels correspond to MGMT unmethylated class. B, An example of voxelwise segmentation for a tumor with an unmethylated MGMT promoter. The sharp borders visible between methylated and unmethylated types result from the patch-wise classification approach.
Training and Segmentation Times
Fine-tuning the network took approximately 1 week. The trained network took approximately 3 minutes to segment the whole tumor and determine the MGMT status for each subject.
DISCUSSION
We developed a fully-automated, highly accurate, deep learning network for determining the methylation status of the MGMT promoter that outperforms previously reported algorithms.33⇓-35 Our network is able to determine MGMT promoter methylation status from T2WI alone. This eliminates potential failures from image-acquisition artifacts and makes clinical translation straightforward because T2WI is routinely obtained as part of standard clinical brain MR imaging. Previous approaches have required multicontrast input, which can be compromised due to patient motion from lengthier examination times and the need for gadolinium contrast. Obviating the need for intravenous contrast makes our algorithm applicable to patients with contrast allergies and renal failure. Compared with previously published algorithms, our methodology is fully automated and uses minimal preprocessing. The time required for the MGMT-net to segment the whole tumor and predict the MGMT promoter methylation status for 1 subject is approximately 3 minutes on a K80 or P40 NVIDA-GPU.
Other groups have also proposed deep learning methods for noninvasive, image-based MGMT molecular profiling, but each of these has several limitations. Korfiatis et al9 implemented a 2D-based slice-wise network, pre-selecting only cases of glioblastoma multiforme for training and prediction. While they achieved a high slice-wise accuracy, their average subject-wise MGMT prediction accuracy was only 90%. Most important, in clinical practice, the tumor grade is unknown a priori. Thus, the approach of Korfiatis et al is a nonviable clinical method from the outset. Our approach of using a mix of low-grade and high-grade gliomas is a better approximation of the real-world clinical workflow in which tissue is not yet available.
Similar to the work of Korfiatis et al, Chang et al35 also implemented a 2D-network, but instead used a case mix like ours (low-grade and high-grade gliomas from the TCIA/TCGA). However, they were only able to achieve an MGMT prediction accuracy of 83% (range, 76%–88%), and their network required tumor presegmentation. Our algorithm far outperformed the approach of Chang et al on a similar dataset without the need for presegmentation. Additionally, it is unclear whether 2D algorithms of either Korfiatis et al9 or Chang et al35 addressed the issue of “data leakage.”28,29 This is a potentially significant limitation for 2D networks that can occur during the slice-wise randomization process if different slices of the same tumor from the same subject are mixed among training, validation, and testing datasets. Unless this is explicitly addressed during the slice-randomization procedure, the reported accuracies can be upwardly biased. Our approach outperforms all prior reports on noninvasive determination of MGMT status and is the first to achieve tissue-level performance, representing a milestone in the clinical viability of MR imaging–based MGMT promoter status prediction.
The higher performance achieved by our network compared with previous image-based classification studies can be explained by several factors. The dense connections in our 3D network architecture are easier to train, carry information from the previous layers to the following layers, and can reduce over-fitting.36,37 3D networks also interpolate between slices to maintain interslice information more accurately. The Dual Volume Fusion postprocessing step improved the Dice scores by approximately 6% by eliminating extraneous voxels not connected to the tumor. Our approach also uses voxelwise classifiers and provides a classification for each voxel in the image. These steps provide simultaneous single-label tumor segmentation. The cross-validation single-label whole-tumor segmentation performance for the MGMT network provided excellent Dice scores of 0.82 [SD, 0.008].
The ability to determine MGMT promoter methylation status on the basis of MR images alone is clinically significant because it helps determine whether the glioma will be susceptible to temozolomide (TMZ). Alkylating agents such as temozolomide damage DNA by methylating the oxygen at position 6 of the guanine nucleotide (O6-methylguanine). The process by which many DNA repair enzymes remove O6-methylguanine, results in DNA breaks, culminating in cell death. However, MGMT works differently by restoring the normal guanine residue and rescuing the glioma cell. Therefore, MGMT activity leads to resistance to therapy. Methylation of the MGMT promoter leads to inactivation of MGMT and loss of resistance of glioma cells to alkylating agents. The MGMT protein is encoded on the long arm of chromosome 10 at position 26 (10q26). Transcription of the MGMT gene is regulated by several promoters.29
Although incompletely understood, at least 9 specific regions within the promoter's gene determine whether a cell will express or not express MGMT.29 However, some regions have been shown to be more important for loss of MGMT expression.38 In the clinical setting, methods for determining MGMT methylation focus on these regions in the promoter gene. The 4 most prevalent methods to detect MGMT methylation are the following: immunohistochemistry, pyrosequencing, quantitative methylation-specific polymerase chain reaction (PCR), and methylation-specific PCR. Pyrosequencing is considered the theoretic criterion standard but is not readily available, and although it is quantitative, there is no agreement on what cutoff values to use when determining MGMT promoter methylation status.30 Therefore, although it is not quantitative, methylation-specific PCR is the most widely used method.39 Additionally, most centers perform MGMT methylation detection on formalin-fixed or paraffin-embedded tissue specimens. These methods have several limitations. Evaluating multiple different methylation sites is technically challenging on a single tissue specimen.39 Tumor heterogeneity poses a substantial limitation of these methods because sampling bias can lead to inaccurate determinations. The presence of hemorrhage, necrosis, or nonmalignant cells contaminates the specimen.39 Therefore, some institutions mandate that at least 50% of the sample to be analyzed contains tumor cells. Prior to PCR, several tissue-processing steps are required. Bisulfite treatment is the most critical step because it will produce the modified DNA that will be used for PCR; however, it also degrades the amount of DNA available, and incomplete treatment can lead to false-positive results.39 The reported sensitivity and specificity of methylation-specific PCR is 91% and 75%, respectively, while the reported sensitivity and specificity of pyrosequencing is 78% and 90%.32
Our noninvasive, MR imaging–based deep learning algorithm outperformed these methods with a sensitivity and specificity of 96.3% and 91.6%, respectively. The overall determination of MGMT promoter methylation status is based on the majority voxels in the tumor. Given the variability in the cutoff values for pyrosequencing-based detection, we performed a Youden statistical index analysis to determine whether the optimal cutoff for our deep learning algorithm was different from majority voting (>50%). The analysis demonstrated that maximum accuracy, sensitivity, specificity, positive predictive value, and negative predictive value were obtained at an optimal cutoff of 50%, the same as majority voting.
Our algorithm was trained on ground truth obtained from the TCGA data base. TCGA uses the Infinium Methylation Assay (https://www.illumina.com/science/technology/microarray/infinium-methylation-assay.html) to determine MGMT promoter methylation status.40⇓-42 Infinium Methylation Assays are an immunofluorescence method that uses next-generation high-throughput microchip arrays and probes. While these methods have been reported to be more sensitive and specific than the most widely available clinical assays, they require pre-existing probes to detect specific methylation sites.42 The sensitivity and specificity values change depending on the probe and analytic model used to interpret the results.42 The sensitivities for the best probes range from 87.5% to 90.6%, while the specificity is 94.4%.42 The overall accuracy of these probes with an optimized analytic model ranges from 91.24% to 93.6%.34 The accuracy of the commercially available Infinium Methylation Assay with the best analytic model is 92%.34 Our algorithm outperforms this assay with a mean cross-validation testing accuracy of 94.73%. While the algorithm appears to outperform the ground truth, there are additional factors that need to be considered for this dataset. The TCGA data base used very stringent tissue screening before molecular testing, including review of tissue to ensure a minimum of 80% tumor nuclei and a maximum of 50% necrosis with additional quality-control measurements of the extracted DNA and RNA before analyses. Additionally, the MGMT determinations made in the TCGA data base were verified by a secondary test.43 Thus, the reported accuracy of the Infinium Methylation Assay is not necessarily comparable with the accuracy in TCIA/TCGA datasets. It is also possible that the algorithm learns features that allow it to perform better than the single-site tissue-biopsy sample ground truth performance because the algorithm “samples” the entire tumor and learns imaging features that are specific to MGMT mutation.
Tissue-based methods for determining MGMT promoter methylation status remain a complex, multistep process that is susceptible to failure and inaccuracy even after an adequate tissue sample has been obtained. Thus, the ability to determine MGMT promoter methylation status on the basis of routine T2WI alone is highly desirable. Additionally, because our algorithm was trained and evaluated on the multi-institutional TCIA database, it is a better representative of algorithm robustness, real-world performance, and potential clinical use than the previously reported methods.25
The algorithm misclassified 13 cases: Six subjects were misclassified as unmethylated, and 7, as methylated. Despite these misclassifications, our network achieved a mean cross-validation testing accuracy of 94.73%, which is higher than that for the methylation-specific PCR, pyrosequencing (PYR), and Infinium Methylation Assays.42 While these tissue-based methods require an invasive procedure and subsequent tissue processing for at least 48 hours, our deep learning algorithm can segment the entire glioma and determine MGMT promoter methylation status in 3 minutes. The deep learning algorithm can also be fine-tuned to variations in institutional MR imaging scanners, while other tissue-based methods currently lack standardization as mentioned above.
The limitations of our study are that deep learning studies require large amounts of data and the relative number of subjects with MGMT promoter methylation is small in the TCGA database. While the number of subjects may seem small, we used a patch-based algorithm with data augmentation, which provided well over 300,000 samples (patches) for training and validation. Additionally, acquisition parameters and imaging vendor platforms vary across imaging centers that contribute data, though this may also be a regarded as a desirable aspect for the generalizability of the approach. Our current classification approach uses a largest connected component step to limit false-positives. As a consequence, multifocal tumors represent a potential limitation. Despite these caveats, our algorithm demonstrated high accuracy in determining MGMT promoter methylation status approaching tissue-level performance.
CONCLUSIONS
We demonstrate high accuracy in determining MGMT promoter methylation status using only T2WI. This represents an important milestone toward using MR imaging to predict glioma histology, prognosis, and appropriate treatment.
ACKNOWLEDGMENTS
We thank Yin Xi, PhD, a statistician, for help with the receiver operating characteristics and areas under the curve.
Footnotes
This work was supported by National Institutes of Health/National Cancer Institute U01CA207091 (A.J.M., J.A.M.).
Disclosures: Chandan Ganesh Bangalore Yogananda—UNRELATED: Employment: University of Texas Southwestern Medical Center. Baowei Fei—RELATED: Grant: National Institutes of Health, Comments: This research was supported, in part, by the US National Institutes of Health grants (R01CA156775, R01CA204254, R01HL140325, and R21CA231911) and by the Cancer Prevention and Research Institute of Texas grant RP190588.* Ananth J. Madhuranthakam—RELATED: Grant: National Institutes of Health/National Cancer Institute, Comments: U01CA207091.* Joseph A. Maldjian—RELATED: Grant: National Institutes of Health/National Cancer Institute grant*; UNRELATED: Consultancy: BioClinica, Comments: blinded clinical trial reader. *Money paid to the institution.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received June 24, 2020.
- Accepted after revision November 21, 2020.
- © 2021 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}