
Abstract
BACKGROUND AND PURPOSE: The World Health Organization has recently placed new emphasis on the integration of genetic information for gliomas. While tissue sampling remains the criterion standard, noninvasive imaging techniques may provide complimentary insight into clinically relevant genetic mutations. Our aim was to train a convolutional neural network to independently predict underlying molecular genetic mutation status in gliomas with high accuracy and identify the most predictive imaging features for each mutation.
MATERIALS AND METHODS: MR imaging data and molecular information were retrospectively obtained from The Cancer Imaging Archives for 259 patients with either low- or high-grade gliomas. A convolutional neural network was trained to classify isocitrate dehydrogenase 1 (IDH1) mutation status, 1p/19q codeletion, and O6-methylguanine-DNA methyltransferase (MGMT) promotor methylation status. Principal component analysis of the final convolutional neural network layer was used to extract the key imaging features critical for successful classification.
RESULTS: Classification had high accuracy: IDH1 mutation status, 94%; 1p/19q codeletion, 92%; and MGMT promotor methylation status, 83%. Each genetic category was also associated with distinctive imaging features such as definition of tumor margins, T1 and FLAIR suppression, extent of edema, extent of necrosis, and textural features.
CONCLUSIONS: Our results indicate that for The Cancer Imaging Archives dataset, machine-learning approaches allow classification of individual genetic mutations of both low- and high-grade gliomas. We show that relevant MR imaging features acquired from an added dimensionality-reduction technique demonstrate that neural networks are capable of learning key imaging components without prior feature selection or human-directed training.
ABBREVIATIONS:
- CNN
- convolutional neural network
- IDH
- isocitrate dehydrogenase
- MGMT
- O6-methylguanine-DNA methyltransferase
- VASARI
- Visually AcceSAble Rembrandt Images
Diffuse infiltrating gliomas are a heterogeneous group of primary tumors with highly variable imaging characteristics, response to therapy, clinical course, and prognoses. This well-known heterogeneity is, in part, attributed to the multiple variations in the genetic and epigenetic mutations that occur early in tumorigenesis.1 For example, isocitrate dehydrogenase 1 and/or 2 (IDH1 and/or 2)–mutant glioblastomas demonstrate significantly improved survivorship compared with IDH wild-type glioblastomas (31 months versus 15 months).2,3 Similarly, patients with anaplastic oligodendrogliomas with 1p/19q codeletion benefit from combined procarbazine/lomustine/vincristine therapy and radiation therapy compared with patients without the mutation.4,5 Regarding chemotherapy response, glioblastomas with O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation demonstrate improved response to the combination of temozolomide and radiation therapy versus radiation therapy alone (21.7 versus 15.3 months).6 The World Health Organization has recently placed new emphasis on the integration of genetic and molecular information for CNS tumor-classification schemes, including IDH1 status and 1p/19q codeletion and several other molecular or genetic markers.7 Thus, knowledge of tumoral genetic information is needed to accurately monitor patients with gliomas and guide personalized therapies.
At present, information regarding underlying genetic and molecular alterations of gliomas is based on analysis of tumor tissue obtained during an operation. However, although high-grade gliomas are known to infiltrate widely into the surrounding nonenhancing peritumoral region,8 biopsies are often limited to the easily accessible areas of the enhancing tumor. Additionally, molecular genetic testing can be costly or not widely available, and the results may take weeks, thereby delaying important therapeutic decisions. Noninvasive imaging techniques that can provide complimentary insight into clinically relevant genetic mutations may expedite and coordinate care among clinicians, minimizing these delays.
MR imaging can noninvasively assess the entire tumor, allowing both a global and regional (voxelwise) characterization of molecular genetics, in contrast to the spatially limited assessment of tissue biopsy. Specifically, both spatial and temporal variations in genetic expression are known to result in heterogeneous alterations in tumor biology, including changes in angiogenesis, cellular proliferation, cellular invasion, and apoptosis.9 These biologic changes are reflected in the complex imaging features of gliomas, manifest by varying degrees of enhancement, infiltration, hemorrhage, reduced diffusion, edema, and necrosis. Attempts to standardize visual interpretation of malignant gliomas for tissue classification have led to the Visually AcceSAble Rembrandt Images (VASARI) feature set, a rule-based lexicon to improve the reproducibility of interpretation.10 However, a limitation of such approaches is the need for a priori feature selection and human visual interpretation, which innately distills a complex dataset of over a million voxels per MR imaging to a handful of numeric descriptors—a “big data” challenge.
The purpose of this study was to classify genetic variations of diffuse infiltrating gliomas using deep-learning/machine-learning approaches implemented with convolutional neural networks (CNNs). CNN approaches model the animal visual cortex by applying a feed-forward artificial neural network to simulate multiple layers of neurons organized in overlapping regions within a visual field, with each layer acting to transform the raw input image into more complex, hierarchic, and abstract representations.11 Thus, it is natural to consider applying deep-learning methods to biomedical images. We hypothesized the following: 1) A CNN can be trained to independently predict underlying molecular genetic mutation status in gliomas with high accuracy, and 2) a trained CNN can identify predictive imaging features for a given mutation.
Materials and Methods
Subjects
MR imaging data were retrospectively obtained from The Cancer Imaging Archives for patients with either low- or high-grade gliomas.12 Corresponding molecular genetic information was obtained from The Cancer Genome Atlas. Only patients with full preoperative MR imaging, including T2, FLAIR, and T1-weighted pre- and postcontrast acquisitions, were included in the analysis. Corresponding molecular information for each patient was obtained, including IDH1 status, 1p/19q codeletion, and MGMT promoter methylation.13
Image Preprocessing
For each patient, all imaging modalities were coregistered using the FMRIB Linear Image Registration Tool (FLIRT; http://www.fmrib.ox.ac.uk/fsl/fslwiki/FLIRT).14,15 Registration was implemented with a linear affine transformation algorithm using 12 df, trilinear interpolation, and a mutual-information cost function. The reference volume for coregistration was the highest resolution sequence, most commonly the postcontrast T1-weighted acquisition. The average time for coregistration was approximately 1 minute per volume. On a typical multicentral processing unit core workstation, the required total of 3 registrations per patient can be performed simultaneously as separate processes, thus allowing all modalities to be coregistered in approximately 1 minute.
Each input technique was independently normalized using z-score values (μ = 0, σ = 1). From these, a custom in-house fully automated whole-brain extraction tool–based 3D convolutional neural network was used to remove extracranial structures. Next, a fully automated brain tumor segmentation tool was used to identify lesion margins. This algorithm was the top-performing tool as evaluated in the international 2016 Multimodal Brain Tumor Segmentation Challenge.16 It is based on a serial fully convolutional neural network architecture with residual connections and performs whole-tumor segmentation in approximately 1 second. These masks were used to generate cropped slice-by-slice images of the tumor on all modalities, each of which were subsequently resized to a 32 × 32 × 4 input.
No other form of preprocessing was necessary for this study. Specifically, the flexibility of CNNs allows robust classification, even in the absence of conventional image-preprocessing steps such as histogram normalization or bias field correction.
Convolutional Neural Networks
CNNs are an adaption of the traditional artificial neural network architecture whereby banks of 2D convolutional filter parameters and nonlinear activation functions act as a mapping function to transform a multidimensional input image into a desired output.17 Network overview and details are provided in the On-line Appendix.
Feature Analysis
The final feature vector produced by a neural network through serial convolutions often encodes for redundant information, given the flexibility of the algorithm to choose any feature necessary to produce accurate classification. In the architecture used for this study, this means that many of 64 features in the final hidden layer will be highly correlated to each other. To decompose this encoded information and gain insight into features learned by the algorithm, we applied principal component analysis to the final feature vector with various dimensionally reduced subspaces, L ϵ.1,5 By means of this approach, the principal component analysis–reduced features, whose weights have the largest absolute value magnitude with respect to the final classification, can be identified. These features can be interpreted as those automatically learned by the algorithm that are most influential in classification of any given mutation status. These final imaging features identified by the algorithm are shown in Figs 1⇓⇓–4.
Statistical Analysis
To evaluate overall per-patient accuracy, we pooled mean softmax scores across all image slices, with a threshold of 0.5 used to determine mutation classification. As an example of IDH status, an average softmax score of >0.5 represents prediction of mutant status, while an average softmax score of <0.5 represents prediction of wild-type status. While a softmax score of 0.5 is the standard threshold for neural network classification, it is possible to arbitrarily change this cutoff to between 0 and 1 to alter the sensitivity and specificity of the network. By means of this approach, overall algorithm performance on a wide range of thresholds is reported as an area under the curve calculation for each mutation.
To assess algorithm generalization, we used a 5-fold cross-validation approach. For each of the 5 experimental iterations, 20% of the data were used for validation, while the remaining 80% of the data were used for training. Results are reported for only patients in the validation cohort after 5-fold cross-validation.
Results
Subjects
A total of 5259 axial slices of tumor from 259 patients with gliomas (135 men/122 women/2 unknown; mean age, 53.2 years; mean survival, 18.8 years) were included for analysis. The distribution of gliomas included the following: 21.2% (55/259) grade II, 22.8% (59/259) grade III, and 55.2% (143/259) grade IV gliomas. There were 2 gliomas that had available genetic information but no World Health Organization grade assignment. IDH1 mutant and wild-type tumors accounted for 45.9% (119/259) and 54.1% (140/259) of patients, respectively; 1p/19q codeletions and nondeletions accounted for 12.0% (31/259) and 88.0% (228/259) of patients, respectively. MGMT promoter methylated and unmethylated accounted for 56.4% (146/259) and 43.6% (113/259) of patients, respectively. The mean tumor size determined by automated segmentation masks was 105.6 cm3.
CNN Accuracy
Overall, the algorithm correctly predicted IDH1 mutation (mean, 94%; range between cross validations, 90%–96%), 1p/19q codeletion (mean, 92%; range, 88%–95%), and MGMT promoter methylation (mean, 83%; range, 76%–88%) on 5-fold cross-validation. The area under the curve for IDH mutation (mean, 0.91; range, 0.89–0.92), 1p/19q codeletion (mean, 0.88; range, 0.85–0.90), and MGMT promoter methylation (mean, 0.81; range, 0.76–0.84) also reflected high performance. The CNN was trained for 25,000 iterations (approximately 3000 epochs with batch sizes ranging from 12 to 48) before convergence. A single forward pass during test time for classification of new cases can be achieved in 0.0043 seconds. Overall, the imaging workflow takes 5.12 seconds per patient (4–5 seconds for detection and preprocessing and the remaining 0.0043 seconds for classification).
Feature Analysis
For IDH1 mutation (Figs 1 and 2), the most predictive features were the following: absent or minimal areas of enhancement (the presence of a larger portion of nonenhancing tumor), central areas of cysts with low T1 and FLAIR suppression, and well-defined tumor margins. By comparison, IDH1 wild-type tumors tended to demonstrate a larger portion of enhancing tumor with thick enhancement; thin, irregular, poorly marginated peripheral enhancement with central necrosis; and an infiltrative pattern of edema (seen as more irregular and ill-defined margins of T2/FLAIR signal abnormality).

T1 postcontrast features most highly correlated with IDH1 mutation status. Prototypical cases as identified by our convolutional neural network imaging features associated with IDH wild types (A and C) and mutation (B and D). Specifically, IDH wild types demonstrate thick and irregular enhancement (A) or thin, irregular poorly marinated peripheral enhancement (B). In contrast, patients with IDH mutation demonstrate absent or minimal enhancement (C) with well-defined tumor margins (D).

FLAIR features most highly correlated with IDH1 mutation status. Prototypical cases as identified by our convolutional neural network imaging features are associated with IDH wild types (A and B) and mutation (C and D). IDH wild types demonstrate infiltrative patterns of edema, seen as more irregular (A) and ill-defined (D) margins of FLAIR signal abnormality. Patients with IDH mutation demonstrate central areas of cysts with FLAIR suppression (C) and well-defined tumor margins (D).
For 1p19 codeletion (Fig 3), the most predictive features were left frontal lobe location, ill-defined tumor margins, and larger portion of enhancement. Compared with either IDH1 mutation or MGMT promoter methylation, many features learned by the CNN for 1p19 codeletion were highly correlated to each other; this finding resulted in an overall smaller number of differentiable features.

MR imaging features most highly correlated with 1p/19q codeletion. Prototypical cases as identified by our convolutional neural network imaging features are associated with 1p/19q absence (A and B) and presence (C and D) codeletion status. Features predictive of the absence of 1p/19q codeletion are poor enhancement (A) and increased vasogenic edema (B). Features predictive of the presence of 1p/19q codeletion are increased enhancement (C) and a left frontal predominance and ill-defined FLAIR margins with mass effect (D).
For MGMT promoter methylation (Fig 4), the most predictive features were a heterogeneous, nodular enhancement; the presence of an eccentric cyst; more masslike edema (larger lesions with a higher portion of nonenhancing tumor component) with cortical involvement; and slight frontal and superficial temporal predominance. By comparison, unmethylated tumors tended to demonstrate thin rim enhancement, central areas of necrosis, solid enhancement, more vasogenic edema, and a slight, deep temporal predominance.
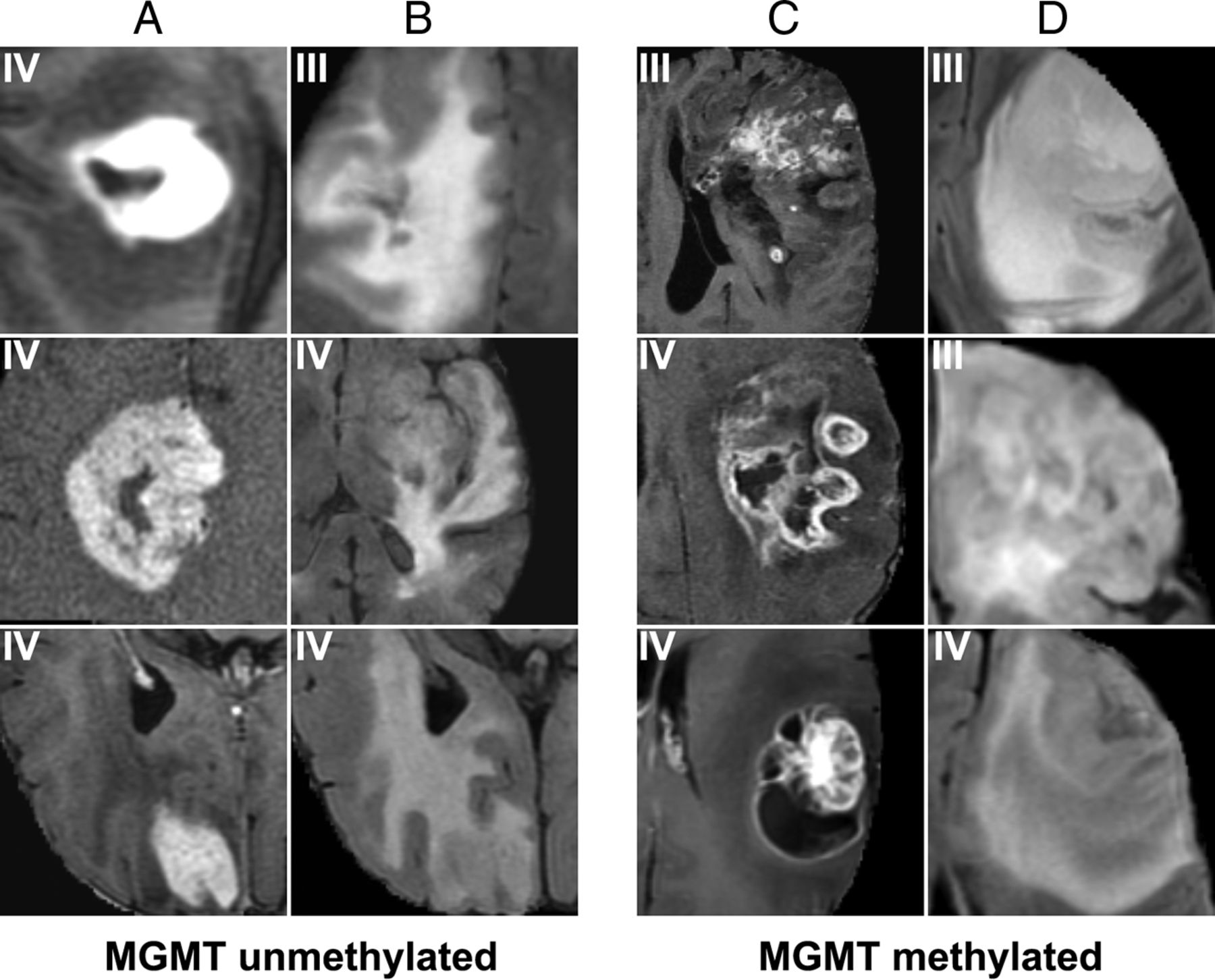
Features most highly correlated with MGMT methylation. Prototypical cases as identified by our convolutional neural network imaging features associated with unmethylated (A and B) and methylated (C and D) MGMT statuses. Features predictive of MGMT unmethylated status include thick enhancement with central necrosis (A) with infiltrative edema patterns (B). In contrast, features predictive of MGMT promoter methylated status include nodular and heterogeneous enhancement (C) with masslike FLAIR edema (D).
Discussion
The variability of clinical outcomes in patients with diffuse infiltrating gliomas is, in part, predicated on molecular and genetic heterogeneity, which has spurred the development and study of noninvasive tools to better classify these tumors. In this study, we used a deep-learning approach to classify individual somatic mutations of diffuse infiltrating gliomas, World Health Organization II–IV, including IDH1 status; 1p/19q codeletion; and the presence of MGMT promoter methylation with an accuracy of 94% (90%–96%), 92% (88%–95%), and 83% (76%–88%), respectively. We were able to implement the entire preprocessing pipeline from tumor detection to tissue segmentation to mutation classification without human supervision. Furthermore, neural networks have been criticized for being “black boxes” that generate uninterpretable feature vectors, which limits insight into the underlying mechanism for image classification. In this study, we applied a dimensionality-reduction approach to visually display the highest ranking features of each mutation category.
Molecular analysis of tumors has significantly impacted the diagnosis of glial tumors, with important implications for both prognosis and therapy guidance. The recently published 2016 World Health Organization Classification of Tumors of the Central Nervous System included several molecular genetic alterations as important features of tumor classification.7 One of the significant changes has been in the classification of oligodendrogliomas, in which mutations of IDH1 or 2 and 1p/19q codeletion are the defining and diagnostic markers. Additionally, hypermethylation of the MGMT promoter, an enzyme involved in DNA de-alkylation and mediation of DNA damage, is a positive prognostic factor.6 Patients with a methylated MGMT promoter have improved survival and better response to radiation therapy with concurrent temozolomide.6,18
Prior classic machine-learning approaches for linking imaging features to these genetic alternations in gliomas have typically relied on human-derived feature extraction such as textural analysis approaches or rule-based systems such as VASARI. For example, Ryu et al19 applied texture analysis to evaluate glioma heterogeneity to distinguish low- and high-grade gliomas with an 80% accuracy. Additionally, Drabycz et al20 described a textural analysis approach to classifying MGMT promoter methylation status in patients with glioblastomas with 71% accuracy. More recently, Kanas et al21 achieved a 74% accuracy in distinguishing the MGMT promoter methylation status from gliomas acquired from The Cancer Genome Atlas using a multivariate prediction model based on qualitative imaging features from the VASARI lexicon. While these approaches have improved the reproducibility and accuracy of classification, the need for manual a priori feature selection remains an inherently limiting factor, a process dependent on expert opinion and an assumption of relevant features.22
As a result, there has been a recent paradigm shift toward end-to-end machine learning using CNNs, which are rapidly outperforming conventional benchmarks on various computer vision tasks.11,23 These models are capable of automatically identifying patterns in complex imaging datasets, thus combining both feature selection and classification into 1 algorithm and removing the need for direct human interaction during the training process. With deep-learning approaches, classification error rates of popular computer vision benchmarks have been significantly lower and now outperform humans on the same task.24⇓–26
Recent use of CNNs has started to yield promising results in multiple medical imaging disciplines, including the detection of pulmonary nodules,27 colon cancer,28 and cerebral microbleeds.29 For example, Lakhani and Sundaram30 applied a CNN approach to automatically identify patients with pulmonary tuberculosis with an area under the curve of 0.99, allowing radiologists to achieve a 97% sensitivity and 100% specificity. This outcome is in comparison with an area under the curve of up to 0.84 using classic machine-learning approaches such as texture and shape analysis.31 Additionally, Chang et al32 developed a CNN approach to automatically identify and count tumor cells from localized biopsy samples of patients with glioblastomas with an accuracy of 96.2%. Zhang et al33 observed that a CNN approach performed significantly better than other techniques for brain segmentation in infants, including random forest, support vector machine, coupled level sets, and most voting.
Given the potential advantages of deep learning, a few studies have also started to explore the use of CNN-based approaches in the determination of glioma mutation status from MR imaging. Recently, Chang et al34 used a 34-layer residual neural network to predict IDH status with up to 89% accuracy using MR imaging in combination with patient age. Compared with the current study, the network used by Chang et al has several million parameters (>1 order magnitude larger than the customized network used in this study), in part limiting overall accuracy through compensatory measures needed to prevent overfitting. Furthermore, only several prototypical slices of the tumor were used (compared with the entire volume in this study), which were then combined in all 3 orthogonal planes (requiring high-resolution isotropic imaging). Korfiatis et al35 also recently described a 50-layer residual network architecture to predict MGMT status. However, the reported classification accuracy of 94.9% comprising 2027 of 2612 images (78%) used for testing contained no tumor at all. Furthermore, the 155 patients used in that study were derived completely from just a single academic center. Finally, in comparison with these prior works, the current study is the first to demonstrate the feasibility of a single neural network architecture to simultaneously predict the status of multiple different mutations (IDH1 status, 1p/19q codeletion, MGMT promoter methylation) with minimal preprocessing in an efficient, fully automated approach.
Despite high accuracy, a commonly cited limitation of CNNs is the apparent difficulty in understanding the underlying black box analytic engine of a network. Several recent studies, however, have proposed novel techniques such as deconvolutional neural networks and occlusion saliency maps to develop a deeper mechanistic understanding of the classification process.36 In this study, we introduced a new technique to visualize the imaging features most relevant to the classification of genetic mutation status using principal component analysis as a means of dimensionality reduction and disentanglement of the final feature vector layer. This approach is useful in medical imaging domains in which the differentiating characteristics of the various disease classes may not be well-established, helping to identify clusters of imaging findings that can be used to guide practicing physicians (Figs 1⇑⇑–4).
In general, the clusters of imaging features identified by the neural network in this study represent a composite of various qualitative descriptions found elsewhere in the literature. For example, MR imaging features predictive of IDH1 mutant status included absent or minimal areas of enhancement, central areas of cystlike necrosis with low T1 and FLAIR suppression, and well-defined tumor margins. This result is in line with existing literature, in which IDH1 mutants have been reported to demonstrate absent or minimal enhancement37⇓–39 and well-defined tumor margins.38,40 By contrast, we observed that IDH1 wild-type tumors demonstrated thick and irregular enhancement with an infiltrative pattern of edema.
For 1p/19q codeletion, the most predictive features were frontal lobe location, ill-defined tumor margins, and increased enhancement. This finding is also in line with existing literature, which has demonstrated that tumors with 1p/19q codeletion are more likely to be found in the frontal cortex.41 Additionally, Sonoda et al39 demonstrated that codeleted tumors are more likely to show contrast enhancement. Finally, the margins of 1p/19q codeleted tumors have also been characterized as poorly circumscribed.42
With regard to MGMT promoter methylation, the most predictive features were a mixed, nodular enhancement; the presence of an eccentric cyst or area of necrosis; more masslike edema with cortical involvement; and slight frontal and superficial temporal predominance. Existing literature has similarly observed that tumors with MGMT promoter methylation tend to have a frontal lobe location43,44 (often colocalization with the IDH1 mutation in this region43) and the presence of an eccentric necrotic cyst.20,45 By comparison, we observed that nonmethylated tumors tended to demonstrate rim enhancement with central areas of necrosis. This observation is also congruent with other literature that has used subjective visual assessment, in which nonmethylated tumors are observed to demonstrate either ring enhancement with central necrosis,20,46 solid enhancement,21 and ill-defined margins.45
When one interprets the results of our study, several limitations should be kept in mind. First, this is a relatively small sample size (n = 259) compared with the neural network studies within the nonmedical domains, which typically include tens of thousands. To address this limitation, we designed a tailormade neural network architecture with a relatively small number of parameters/layers and high normalization. Additionally, all imaging input was resampled to a relatively small size (32 × 32 × 4) to prevent overfitting. Therefore, input for prediction is limited to 4096 voxels on any given slice of tumor as opposed to the potential tens of thousands of voxels. Second, this study is a retrospective study of The Cancer Imaging Archives dataset, a heterogeneous dataset from multiple different contributing sites. However, the success of our network on this dataset suggests that the underlying CNN approach in this study is capable of handling nonuniform imaging protocols. Last, this study is limited by lack of an independent dataset. While the cross-fold validation technique used in this study ensures that the model generalizes well to held-out cohorts from The Cancer Imaging Archives dataset, generalization to unseen datasets remains to be determined. Future studies will need to expand the training set to include a variety of cancer sites and MR imaging scanners.
Conclusions
The results of our study show the feasibility of a deep-learning CNN approach for the accurate classification of individual genetic mutations of both low- and high-grade gliomas. Furthermore, we demonstrate that the relevant MR imaging features acquired from an added dimensionality-reduction technique are concordant with existing literature, showing that neural networks are capable of learning key imaging components without prior feature selection or human directed training.
Footnotes
Disclosures: Peter Chang—RELATED: Grant: National Institutes of Health (National Institute of Biomedical Imaging and Bioengineering) T32 Training Grant, T32EB001631*. Christopher G. Filippi—UNRELATED: Consultancy: KOL Philips Healthcare, Comments: part of a Key Opinion Leaders consortium in which I have advocated for CNNs in advanced imaging including neoplasms; Payment for Lectures Including Service on Speakers Bureaus: Visiting Professor, Comments: In a talk on advanced imaging of tumor, some of the preliminary work was included. Pierre Baldi—RELATED: Grant: National Institutes of Health*; UNRELATED: Royalties: MIT Press, Cambridge University Press, Wiley. Daniel Chow—RELATED: Grant: funding support from Canon Medical Systems USA. *Money paid to the institution.
This work was supported by Canon Medical Systems USA. The work of P.B. was supported, in part, by the following grants: Defense Advanced Research Projects Agency D17AP00002 and National Institutes of Health GM123558. The work of P.C. was in part supported by the National Institutes of Health training grant T32EB001631.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received November 29, 2017.
- Accepted after revision March 20, 2018.
- © 2018 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Integration of MRI radiomics and germline genetics to predict the IDH mutation status of gliomas
- Imaging Genomics of Glioma Revisited: Analytic Methods to Understand Spatial and Temporal Heterogeneity
- Biologically-informed deep neural networks provide quantitative assessment of intratumoral heterogeneity in post-treatment glioblastoma
- Prediction of cancer treatment response from histopathology images through imputed transcriptomics
- Clinical Knowledge and Reasoning Abilities of AI Large Language Models in Anesthesiology: A Comparative Study on the ABA Exam
- IDH and 1p19q Diagnosis in Diffuse Glioma from Preoperative MRI Using Artificial Intelligence
- A Novel Fully Automated MRI-Based Deep Learning Method for Classification of IDH Mutation Status in Brain Gliomas
- Privacy Risks of Whole-Slide Image Sharing in Digital Pathology
- Diffusion Weighted MRI could precisely predict the pTERT mutation status of GBM using a residual convolutional neural network
- Prediction of cancer treatment response from histopathology images through imputed transcriptomics
- Evaluating the Clinical Utility of Artificial Intelligence Assistance and its Explanation on Glioma Grading Task
- Evolving Role and Translation of Radiomics and Radiogenomics in Adult and Pediatric Neuro-Oncology
- Development and Validation of a Deep Learning-Based Model to Distinguish Glioblastoma from Solitary Brain Metastasis Using Conventional MR Images
- MRI-Based Deep-Learning Method for Determining Glioma MGMT Promoter Methylation Status
- Improved Glioma Grading Using Deep Convolutional Neural Networks
- A Combined Deep CNN-LSTM Network for the Detection of Novel Coronavirus (COVID-19) Using X-ray Images
- neoDL: A novel neoantigen intrinsic feature-based deep learning model identifies IDH wild-type glioblastomas with the longest survival
- Neuro-Oncology and Radiogenomics: Time to Integrate?
- Machine learning assisted DSC-MRI radiomics as a tool for glioma classification by grade and mutation status
- Artificial Intelligence in Neuroradiology: Current Status and Future Directions
- A Novel Fully Automated MRI-Based Deep Learning Method for Classification of 1P/19Q Co-Deletion Status in Brain Gliomas
- Brain Tumor IDH, 1p/19q, and MGMT Molecular Classification Using MRI-based Deep Learning: Effect of Motion and Motion Correction
- MRI-BASED DEEP LEARNING METHOD FOR DETERMINING METHYLATION STATUS OF THE O6-METHYLGUANINE-DNA METHYLTRANSFERASE PROMOTER OUTPERFORMS TISSUE BASED METHODS IN BRAIN GLIOMAS
- Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis
- Prediction of Tumor Grade and Nodal Status in Oropharyngeal and Oral Cavity Squamous-cell Carcinoma Using a Radiomic Approach
- Predicting the 1p/19q Codeletion Status of Presumed Low-Grade Glioma with an Externally Validated Machine Learning Algorithm
- Randomised controlled trial of WISENSE, a real-time quality improving system for monitoring blind spots during esophagogastroduodenoscopy
- Transcriptomic learning for digital pathology
- Classification of Brain Tumor IDH Status using MRI and Deep Learning