
Abstract
SUMMARY: This article is the second in a 2-part series reviewing neuroimaging in childhood SNHL. Previously, we discussed the clinical work-up of children with hearing impairment, the classification of inner ear malformations, and congenital nonsyndromic causes of hearing loss. Here, we review and illustrate the most common syndromic hereditary and acquired causes of childhood SNHL, with an emphasis on entities that demonstrate inner ear abnormalities on cross-sectional imaging. Syndromes discussed include BOR syndrome, CHARGE syndrome, Pendred syndrome, Waardenburg syndrome, and X-linked hearing loss with stapes gusher. We conclude the article with a review of acquired causes of childhood SNHL, including infections, trauma, and neoplasms.
ABBREVIATIONS:
- BOR
- branchio-oto-renal
- CISS
- constructive interference in steady state
- IAC
- internal auditory canal
- NF-2
- neurofibromatosis type II
- SCC
- semicircular canal
- SNHL
- sensorineural hearing loss
- T1WI
- T1-weighted image
- T2WI
- T2-weighted image
The estimated prevalence of SNHL in patients younger than 18 years of age is 6 per 1000,1 making it one of the leading causes of childhood disability and a common reason for otolaryngology referrals. Cross-sectional imaging is now routinely performed in these patients because it provides important information about potential etiologies for hearing loss, defines the anatomy of the temporal bone and the central auditory pathway, and identifies additional intracranial abnormalities that may require further work-up. In the first part of our series, we reviewed some of the practical aspects related to imaging children with SNHL, including epidemiology, clinical work-up, and choices of imaging technique; we also discussed the classification of congenital inner ear malformations and congenital nonsyndromic causes of SNHL. In this article, we continue our discussion of neuroimaging for childhood SNHL, focusing on several of the most common syndromic hereditary forms of SNHL as well as acquired causes of hearing loss.
Syndromic Hereditary Causes of Hearing Loss
As was discussed in Part 1, roughly 50% of cases of congenital SNHL can be linked to a genetic cause, with approximately 30% of these considered syndromic and the remaining 70% being nonsyndromic.2 The term “syndromic” implies the presence of other distinctive clinical features in addition to hearing loss, and to date, >300 syndromic forms of hearing loss have been described.3 In many syndromes, hearing loss is an inconstant feature, and a complete review of all syndromes associated with hearing loss is beyond the scope of this review. However, there are a number of well-characterized entities in which SNHL is a frequent and/or major component (Table 1). Although many of these syndromes do not usually demonstrate gross inner ear anomalies by imaging, there are several in which inner ear malformations are a common and sometimes defining feature.
Selected hereditary syndromes commonly associated with SNHL
BOR Syndrome
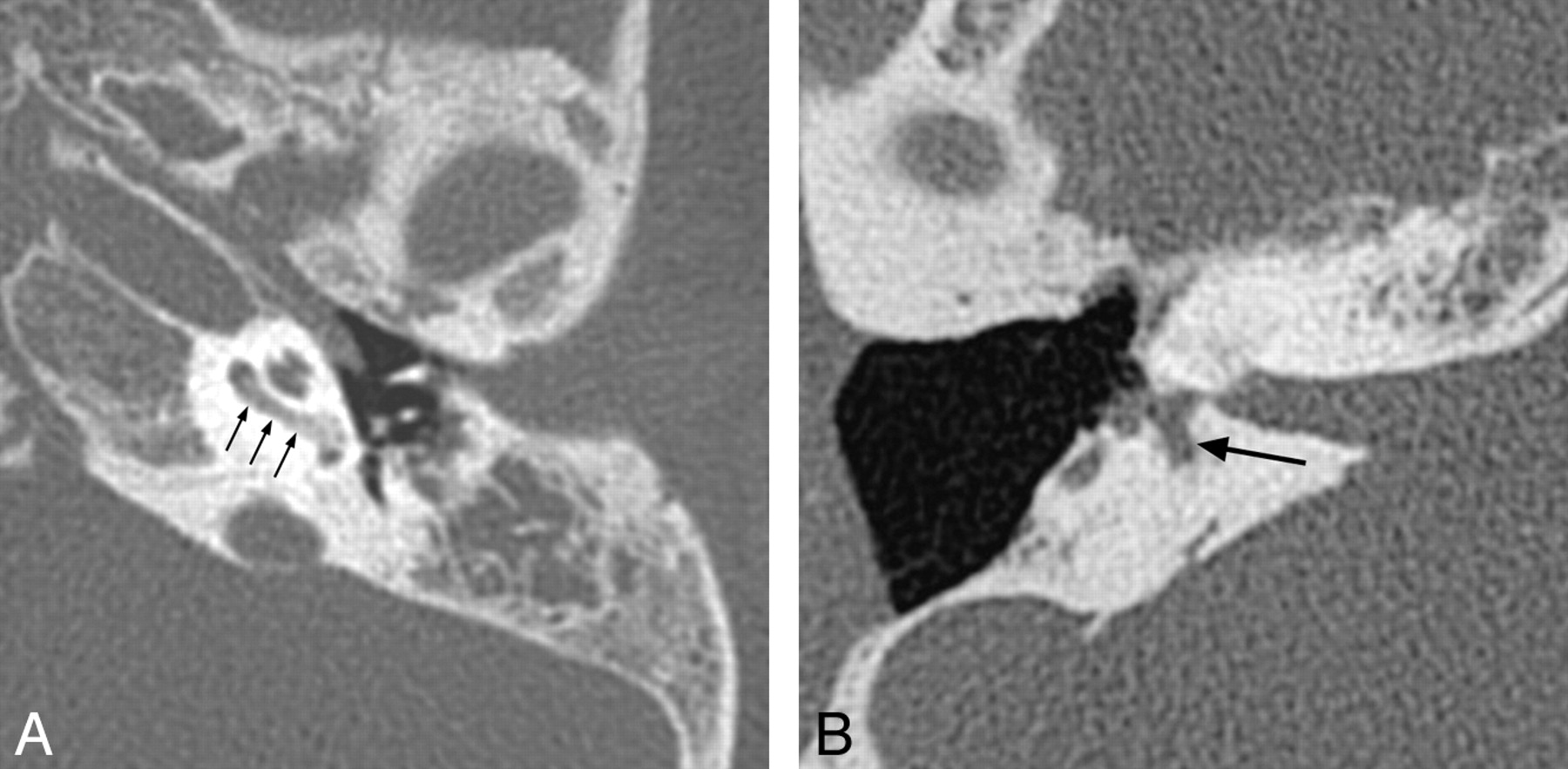
BOR syndrome is autosomal dominant and consists of hearing loss, auricular malformations, branchial arch closure defects (preauricular pits and tags), and renal anomalies. Patients with BOR syndrome may also demonstrate lacrimal duct stenosis, a narrow face, palatal abnormalities, and anomalies of the bladder and ureters. Mutations of 2 genes, EYA1 and SIX1, are known to cause the BOR phenotype, with EYA1 mutations accounting for approximately 40% of cases.4
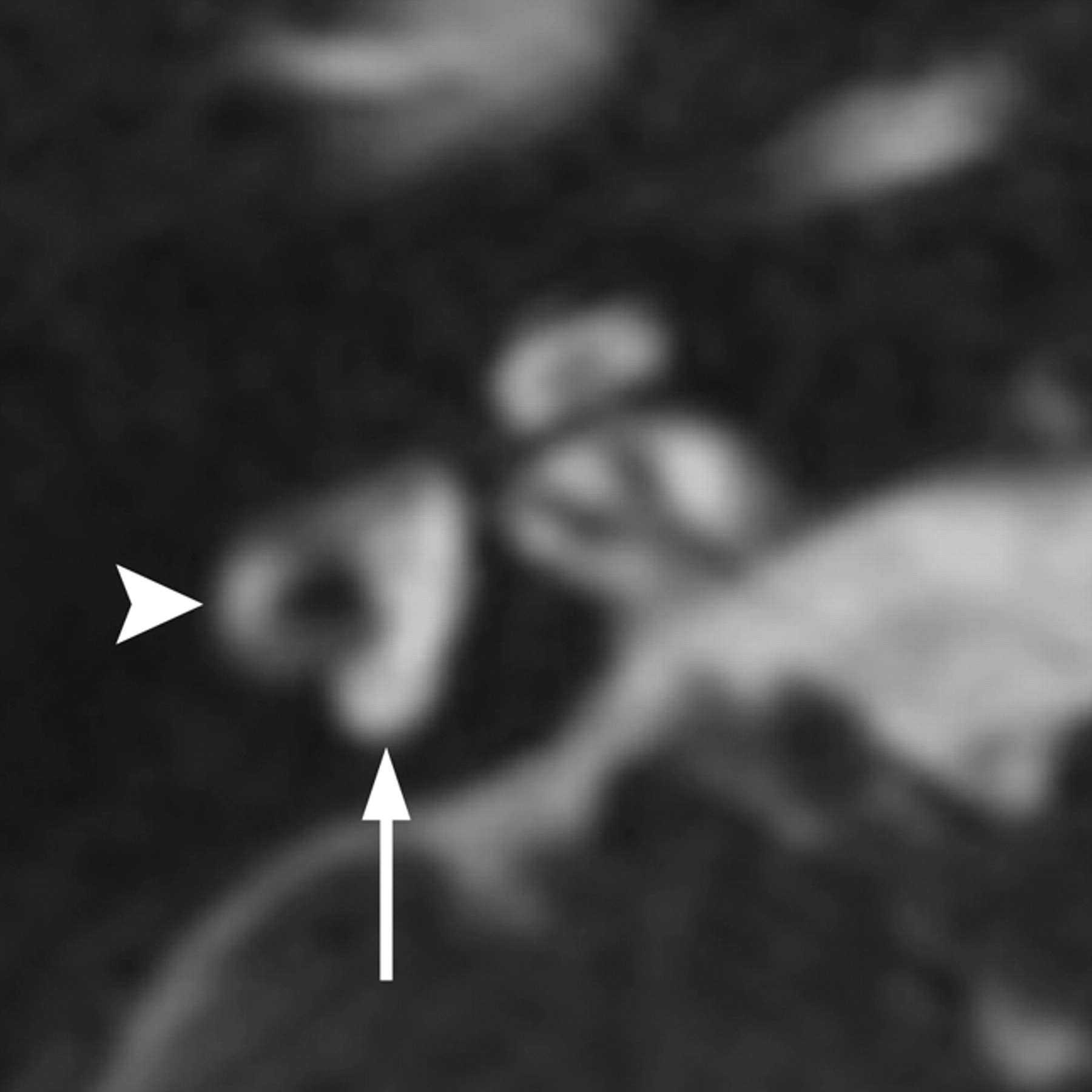
Hearing impairment occurs in 70%–93% of individuals with BOR syndrome, with the age of onset varying from early childhood to young adulthood. Hearing loss may be conductive, sensorineural, or mixed and may range from mild to profound.4 Inner ear anomalies seen in BOR syndrome include cochlear hypoplasia, particularly involving the apical turn; deviation of the labyrinthine facial nerve canal medial to the cochlea (Fig 1); and a funnel-shaped IAC with a large porus acousticus.5 Vestibular dysplasia, SCC hypoplasia, enlargement of the vestibular aqueduct, and cochlear nerve deficiency are also reported. Patients with BOR syndrome also frequently demonstrate stenosis or atresia of the external auditory canal, middle ear and ossicular chain abnormalities (Fig 1), eustachain tube dilation, and absence of the stapedius muscle.5⇓–7

BOR syndrome. A, Axial CT image through the level of the cochlear aperture demonstrates a hypoplastic cochlea without a definable modiolus or apical turn. B, An image slightly superior to A demonstrates a medially displaced canal for the labyrinthine segment of the facial nerve (arrow) and an enlarged vestibular aqueduct. This patient also has a left middle ear cholesteatoma causing ossicular erosion. Only the head of the malleus remains (arrowhead in A). C, CT image through the right epitympanum in the same patient demonstrates a foreshortened dysplastic incus with an enlarged body that essentially articulates directly with the stapes.
CHARGE Syndrome
The CHARGE acronym was originally coined by Pagon et al8 in 1981 and stands for Coloboma, Heart defects, Atresia of the choanae, Retarded growth and development, Genital hypoplasia, and Ear anomalies and/or deafness, reflecting 6 cardinal clinical features of the syndrome. Since its initial description, the definition of CHARGE syndrome has evolved to accommodate several additional anomalies, including rhombencephalic dysfunction, cranial neuropathies, and dysfunction of the hypothalamic-hypophyseal axis. In 2005, Verloes9 updated these criteria, emphasizing the triad of coloboma, choanal atresia, and abnormal SCCs and suggested categories for partial and atypical forms of the syndrome in addition to the typical form (Table 2).
Updated diagnostic criteria for CHARGE syndrome9
CHARGE syndrome is usually a sporadic autosomal dominant disorder, and approximately two-thirds of patients have mutations of the CHD7 gene located on chromosome 8. Deafness is observed in ≤90% of patients, and the hearing loss can be conductive, sensorineural, or mixed.10 Small misshaped pinnae, which are low-set, anteverted, cup-shaped, and widened with reduced vertical height, are present in 95%–100% of patients. Small middle ear cavities, absence of the stapedius muscle, absence of the round and oval windows, hypoplasia of the incus and stapes, ossicular chain fixation, and an abnormal course of the tympanic facial nerve may be seen on CT.11⇓–13
The characteristic inner ear anomaly in CHARGE syndrome is SCC aplasia with associated vestibular dysplasia, which is seen in virtually all patients with the syndrome (Fig 2). Cochlear nerve deficiency with atresia of the cochlear aperture, abnormalities of cochlear partitioning, and anomalies of cranial nerves and olfactory bulbs (best visualized on CISS sequences) are also common.11,13 Bony or membranous choanal atresia may be seen on images through the nasal cavities, and colobomas are frequently evident on images through the orbits (Fig 2).

CHARGE syndrome. A, Axial CISS image through the left temporal bone demonstrates a small vestibule (white arrow) and absence of the lateral SCC. The cochlea is also hypoplastic (arrowhead). B, Axial fast spin-echo T2WI through the brain and orbits demonstrates a left-sided coloboma (black arrow).
Pendred Syndrome
Pendred syndrome is believed to be the most common syndromic form of congenital deafness. It is an autosomal recessive disorder characterized by the combination of euthyroid goiter and severe SNHL and represents between 4.3% and 7.5% of all causes of childhood deafness.14 Goiter in the syndrome usually manifests in midchildhood and is the result of a specific defect in the organification of iodine, which can be a positive demonstrated with a positive perchlorate discharge test.15 The specific mutation causing the syndrome has been mapped to the pds locus on chromosome 7q31.14
Inner ear malformations are almost invariably present on CT and include modiolar deficiency and vestibular enlargement (100%), absence of the interscalar septum between the upper and middle cochlear turns (75%), and enlargement of the vestibular aqueduct (80%).16 Endolymphatic sac enlargement has been reported on MR imaging in 100% of patients with Pendred syndrome.17
Waardenburg Syndrome
Waardenburg syndrome is autosomal dominant with characteristic features, including hypertelorism with a prominent broad nasal root (dystopia canthorum); eyebrow hyperplasia and synophrys; pigmentary disturbances, including heterochromia iridis, a white forelock, leukoderma, and white eyelashes; and SNHL. Several types (I-IV) and additional further subtypes have been identified since the syndrome was initially described, with types I and II occurring most frequently.18,19 Six different genes (PAX3, MITF, EDN3, EDNRB, SOX10, and SNAI2) have been implicated in the different types of this syndrome.19
Hearing loss is the most common feature of Waardenburg syndrome, occurring in approximately 60% of children with type I and 90% of children with type 2. Temporal bone anomalies are reported in ≤50% of patients.18,20 Inner ear abnormalities include vestibular aqueduct enlargement, widening of the upper vestibule, IAC hypoplasia, decreased modiolus size, and, most characteristically, aplasia or hypoplasia of the posterior SCC (seen in roughly 26%) (Fig 3).20,21
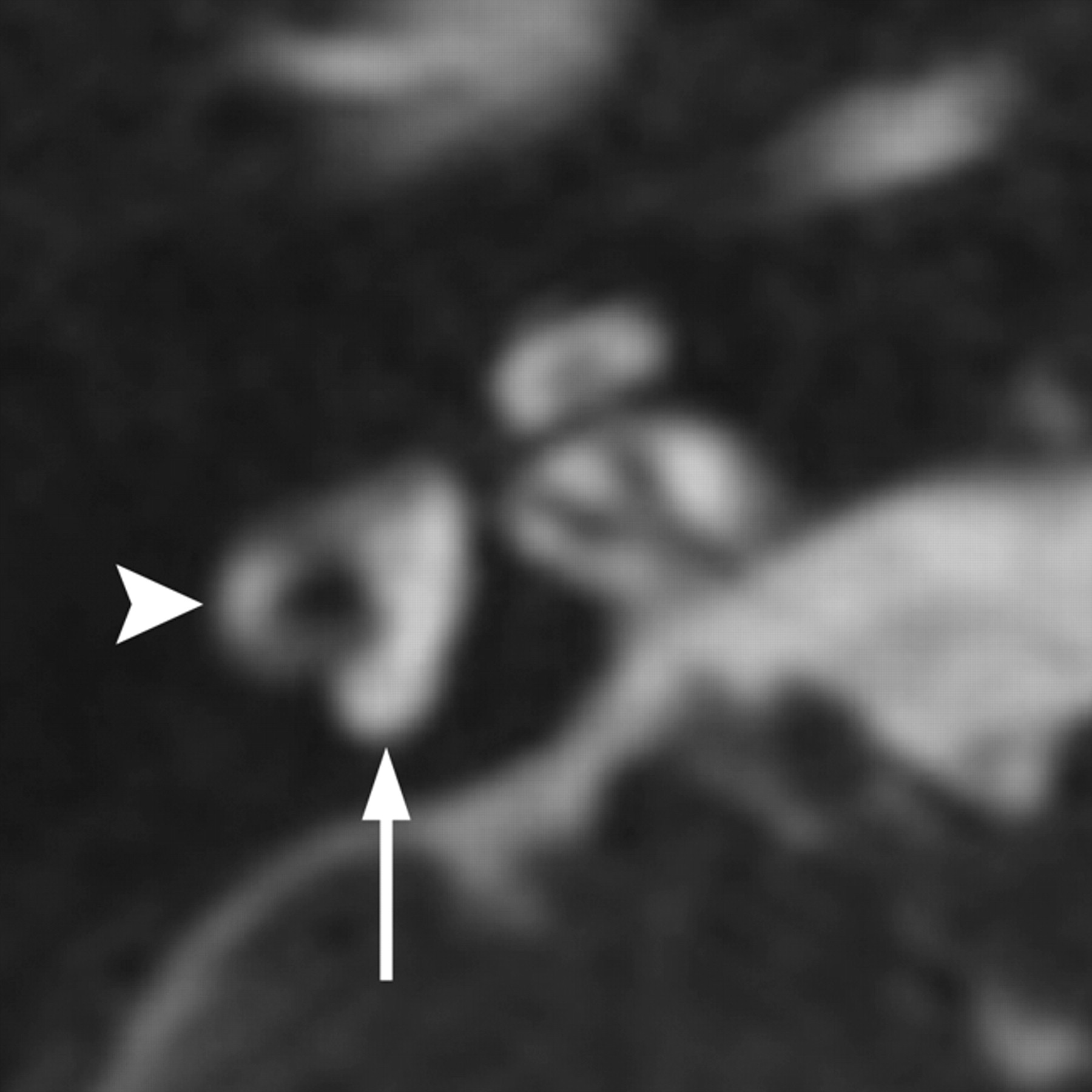
Waardenburg syndrome. Axial CISS image through the right temporal bone demonstrates absence of the posterior SCC (arrow) but a normal lateral SCC (arrowhead). This is a characteristic finding of Waardenburg and Alagille syndromes.
X-Linked Deafness with Stapes Gusher
The syndrome of X-linked deafness with stapes gusher (also known as X-linked deafness type 3, X-linked stapes gusher syndrome, or Nance deafness) is a sex-linked recessive disorder caused by a loss-of-function mutation of the POU3F4 gene at the DFN3 locus of the X chromosome. DFN3 is 1 of 4 X-linked loci implicated in congenital deafness (the others being DF2, DF4, and DF6) and has been found in approximately 50% of families with X-linked hearing loss.22 Symptomatic patients with this mutation are typically male and present with hearing loss at birth, which rapidly progresses to severe deafness within the first decade. Hearing loss is typically mixed, but some individuals demonstrate SNHL without a conductive component. Vestibular problems are also common in these patients.22 Female carriers of the gene mutation may have normal hearing or only mild-to-moderate hearing loss.23
In patients with the DFN3 mutation, there is a communication between the subarachnoid and perilymphatic spaces due to deficiency of the lamina cribrosa separating the IAC from the basal turn of the cochlea. This communication causes elevated perilymphatic pressure and fixation of the stapes footplate, which may result in a conductive hearing loss.24 Manipulation of the stapes during surgery can result in a gush of perilymph through the oval window in these patients (hence the term “stapes gusher”); therefore, stapedectomy is generally contraindicated in these patients.
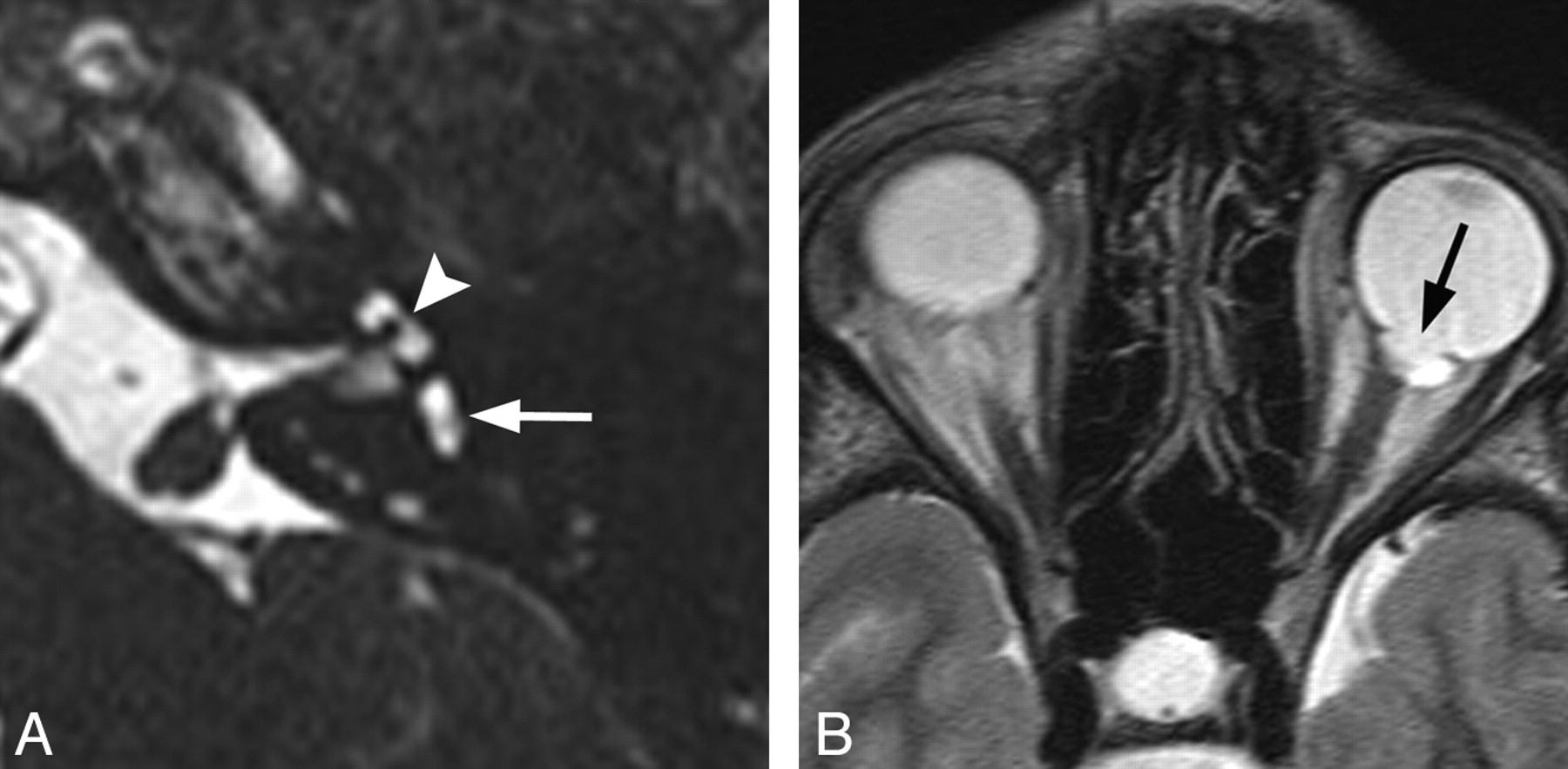
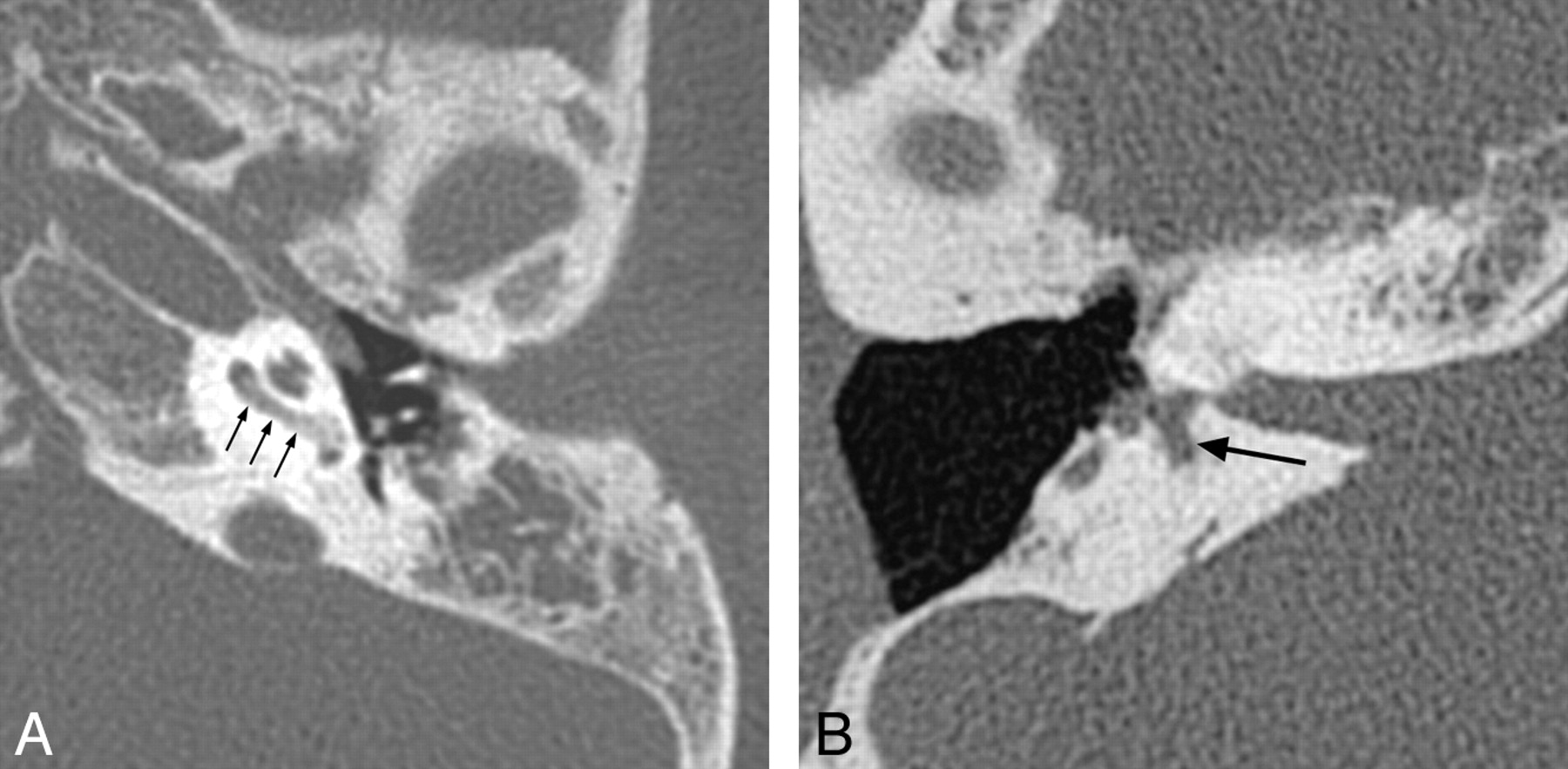
Characteristic findings on both CT and MR imaging in children with X-linked deafness with stapes gusher include enlarged bulbous IACs; a widened cochlear aperture with absence of the lamina cribrosa, appearing as wide communication between the basal turn of the cochlea and the IAC; cochlear hypoplasia with modiolar deficiency; and widening of the bony canal for the labyrinthine segment of the facial nerve (Fig 4).25 Dilation of the vestibular aqueducts has also been reported.26

X-linked hearing loss with stapes gusher. A, Axial CT image through the mid-IAC on the left demonstrates a bulbous IAC, widening of the cochlear aperture, and cochlear hypoplasia with modiolar deficiency. The vestibule is also dilated. B, Axial CT image slightly superior to A demonstrates widening of the bony canal for the labyrinthine segment of the facial nerve (arrow).
Acquired SNHL
Acquired forms of SNHL usually present later in childhood and can be the result of inner ear infections, autoimmune disorders, trauma, or posterior fossa or IAC tumors. Several of these are discussed below.
Infectious Labyrinthitis
Labyrinthitis refers to an inflammatory process of the membranous labyrinth of the inner ear, which typically manifests as acute SNHL or vertigo. Labyrinthitis is classified on the basis of the causative agent and can be considered infectious (bacterial, viral, or luetic) or noninfectious (trauma, autoimmune, or toxic). Infectious labyrinthitis can be further categorized on the basis of its route of spread into the inner ear as tympanogenic, meningogenic, or hematogenic.
In children, meningitis is the most common postnatal cause of acquired bilateral SNHL.27 Meningogenic labyrinthitis is most frequently due to bacterial meningitis and is usually bilateral. The offending pathogens are believed to invade the membranous labyrinth through the cochlear aqueducts or the lamina cribrosa of the vestibule, resulting in a suppurative labyrinthitis.28 In cases of suppurative labyrinthitis due to spread from middle ear infections (tympanogenic labyrinthitis), organisms gain access into the inner ear through the round window, the oval window, or via an anomalous connection between the middle and inner ear. As such, hearing loss in these cases is typically unilateral.
Hematogenic labyrinthitis is less common than either the meningogenic or tympanogenic forms. It is due to seeding from the bloodstream and is most commonly viral in etiology. A number of viruses, including measles, mumps, influenza, rubella, cytomegalovirus, and herpes are associated with acquired inner ear pathology.29,30 Viral labyrinthitis is typically self-limited, and hearing loss may improve or completely resolve before the need for imaging.
Three radiologic stages are described in labyrinthitis: 1) the acute stage, 2) the fibrous stage, and 3) labyrinthitis ossificans. The acute stage is characterized by the presence of inflammatory cells and a serofibrinous exudate in the perilymph with breakdown of the blood-endolymph barrier. MR imaging demonstrates strong labyrinthine enhancement on gadolinium-enhanced T1WI during this stage; however, enhancement is not specific for an infectious etiology because similar findings can be seen with noninfectious causes of labyrinthitis (see below).30 On CT, the inner ear appears normal, though opacification of the involved middle ear and mastoids may be evident in cases of tympanogenic labyrinthitis.
In the fibrous stage of labyrinthitis, which can occur by as early as 2 weeks, granulation tissue consisting of hypertrophic fibroblasts and neovascularity causes membranous fibrosis, which on CISS images is seen as loss of the normal fluid signal intensity in the membranous labyrinth.28 Contrast-enhanced T1WI may demonstrate enhancement of the inner ear, though the enhancement is typically not as strong as that in the acute phase.31 On CT, the inner ear will appear normal, with no evidence of inner ear ossification (Fig 5 ).

Labyrinthine fibrosis. A, Axial CISS image through the left IAC demonstrates loss of normal high-fluid signal intensity in the left cochlea (arrow). B, On the corresponding axial CT image through the left temporal bone, the turns of the cochlea are not ossified; however, subtle thickening of the modiolus may reflect very early cochlear ossification.
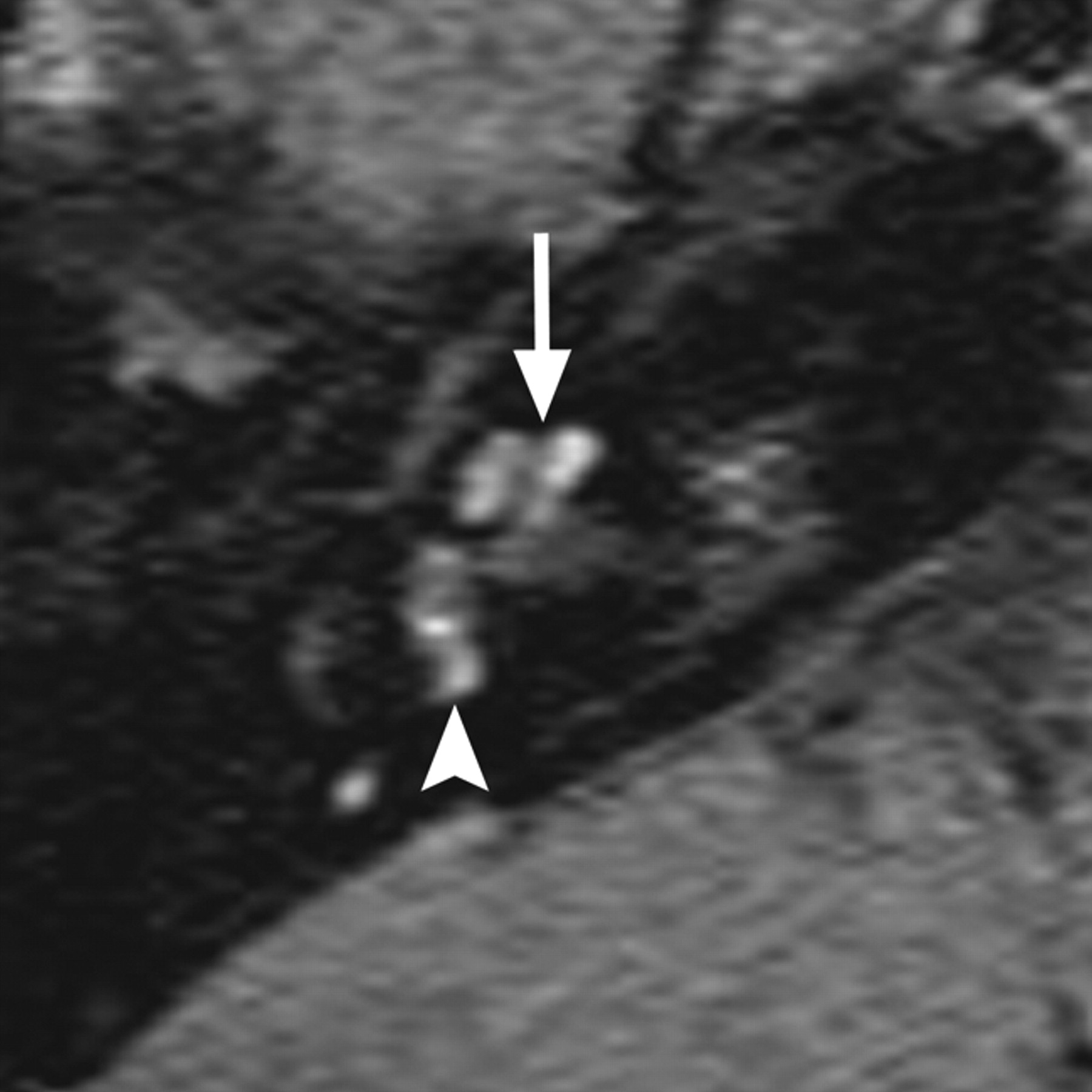
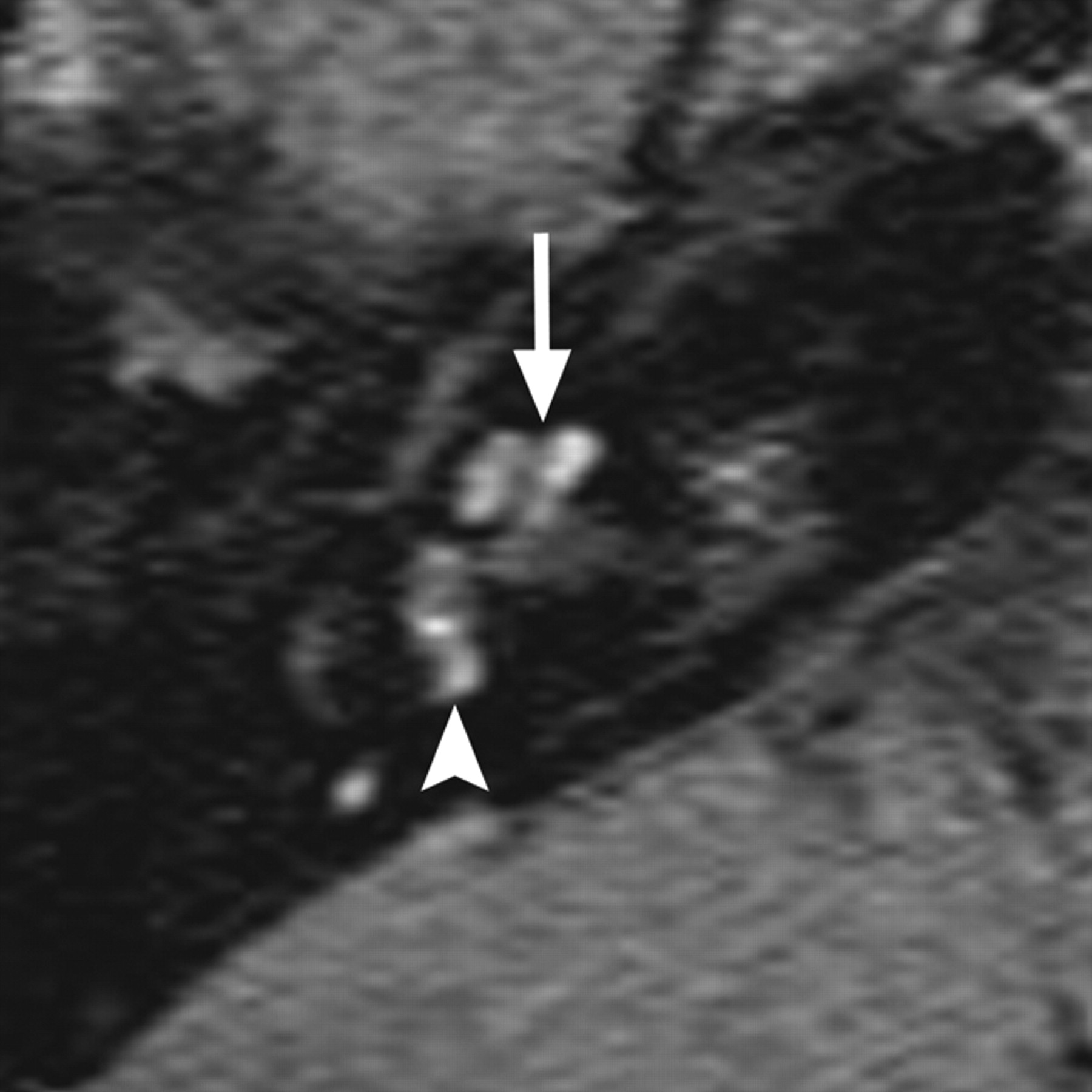
In the final ossifying stage of labyrinthitis, there is metaplastic or osteoplastic bone formation, which appears on CT as calcification within the inner ear (Fig 6 ). Cochlear ossification commonly begins in the basal turn portion of the scala tympani near the round window and progresses toward the apical turn.32,33 MR imaging will demonstrate loss of normal hyperintense fluid signal intensity on CISS images, similar to what is seen in the fibrous stage, without enhancement on contrast-enhanced T1WI.31 As such, fibrosing and ossifying labyrinthitis may be indistinguishable on MR imaging, and CT is necessary to determine the presence of inner ear ossification. The distinction between fibrous and osseous obstruction of the membranous labyrinth is particularly important in candidates for cochlear implantation because significant ossification of the cochlea can make implantation more difficult, if not impossible, and often results in poorer functional results.34
Two cases of labyrinthitis ossificans. A, Axial left temporal bone image demonstrates increased attenuation in the basal turn of the cochlea (small arrows) in a patient with early labyrinthitis ossificans. B, Axial right temporal bone image in a patient who has undergone a previous tympanomastoidectomy demonstrates complete ossification of the cochlea and lateral and posterior SCCs, consistent with advanced labyrinthitis ossificans. Portions of the vestibule (large arrow) remain unossified. Note the normal-sized IAC, which helps to distinguish this from labyrinthine aplasia.
When ossification of the labyrinth becomes very advanced, there may be no recognizable labyrinthine structures, in which case the chief differential consideration is labyrinthine aplasia. Helpful in distinguishing the 2 entities is the fact that in labyrinthine aplasia, the inner ear is typically small with associated flattening of the medial middle ear wall and the IAC is usually hypoplastic or atretic.35
Autoimmune Labyrinthitis
The diagnosis of autoimmune labyrinthitis is usually one of exclusion, and patients typically present with rapidly progressive fluctuating bilateral SNHL that responds to immunosuppressive agents. Vestibular symptoms similar to those seen in Menière disease occur frequently. Supportive laboratory tests include an elevated erythrocyte sedimentation rate, rheumatoid factor, C-reactive protein, or serum immunoglobulins, as well as elevations in non-tissue-specific antibody titers (antinuclear antibody, antineutrophil cytoplasmic antibody, antiendothelial cell antibody). Entities associated with autoimmune hearing loss include Cogan syndrome, systemic lupus erythematosus, juvenile idiopathic arthritis (formerly juvenile rheumatoid arthritis), Wegener granulomatosis, Sjögren syndrome, Behçet disease, antiphospholipid syndrome, anticardiolipin syndrome, and Hashimoto thyroiditis. The prevalence of autoimmune hearing loss increases with age and, in the pediatric population, is highest in late adolescence.36
Autoimmune labyrinthitis appears similar to infectious forms of labyrinthitis on MR imaging and will demonstrate intense labyrinthine enhancement acutely on postcontrast T1WIs (Fig 7 ).28 The imaging abnormalities often resolve with steroid treatment but may occasionally progress to fibrosing or ossifying labyrinthitis.37
Autoimmune labyrinthitis. Axial gadolinium-enhanced T1WI through the right inner ear demonstrates enhancement of the cochlea (arrow), vestibule (arrowhead), and SCCs. Acute infectious labyrinthitis would have an identical appearance.
Trauma
Up to 82% of children with temporal bone fractures will have hearing loss at presentation; of these cases, 56% will be conductive, 17% will be sensorineural, and 10% will be mixed.38 Temporal bone fractures causing SNHL are classically of the transverse variety, which are more likely than longitudinal fractures to involve the bony labyrinth or IAC (Fig 8). Transverse fractures also have a higher incidence of associated facial nerve injury. In reality, although the distinction between transverse and longitudinal fracture orientation is useful for conceptualizing these processes, most temporal bone fractures tend to be complex or oblique in their orientation.39,40

Transverse temporal bone fracture resulting in a perilymph fistula. A, Axial left temporal bone CT image through the level of the superior SCC demonstrates a transversely oriented fracture (arrow), which involves the posterior limb of the superior SCC. B, Axial CT image just inferior to A demonstrates pneumolabyrinth in the vestibule and cochlea. In addition, there is gas in the vestibular aqueduct, which happens to be enlarged in this patient.
Posttraumatic SNHL also occurs in the absence of a temporal bone fracture. In these cases, the injury is referred to as a “cochlear concussion,” a term used to indicate trauma to the membranous labyrinth without a visible fracture. Principal theories proposed to explain the etiology of hearing loss in these patients include the following: 1) disruption of the membranous portion of the cochlea by pressure waves transmitted from CSF, 2) disturbance of cochlear microcirculation, and 3) hemorrhage into the cochlea.41 In these patients, the bony capsule and IAC appear intact on temporal bone CT, but MR imaging may demonstrate hemorrhage in the cochlea or other segments of the membranous labyrinth, which appears as high signal intensity in the inner ear on unenhanced T1WI (Fig 9 ).42 Similar to inflammatory causes of labyrinthitis, hemolabyrinth can progress to labyrinthine ossification.

Hemolabyrinth. Coronal unenhanced T1WI through the cochleae demonstrates increased signal intensity in the right cochlea (arrow), consistent with hemorrhage following trauma.
Perilymph fistulas, which are defined as abnormal communications between the middle and inner ear resulting in leakage of perilymph into the middle ear, can also cause SNHL and vertigo. Most frequently, these fistulas occur in conjunction with temporal bone fractures traversing the middle ear and labyrinth, but they can also occur without a demonstrable fracture. Perilymph fistulas have been reported to occur spontaneously or as a result of mild barotrauma. In these instances, the site of the perilymph leak is usually the round or oval window.43,44 CT in cases of perilymph fistula may demonstrate pneumolabyrinth (Fig 8), but often, the only imaging clue is the presence of a middle ear effusion. Perilymph fistulas also occur as a result of erosion into the membranous labyrinth by a middle ear cholesteatoma or chronic otitis media (most frequently at the apex of the lateral SCC).44
Tumors
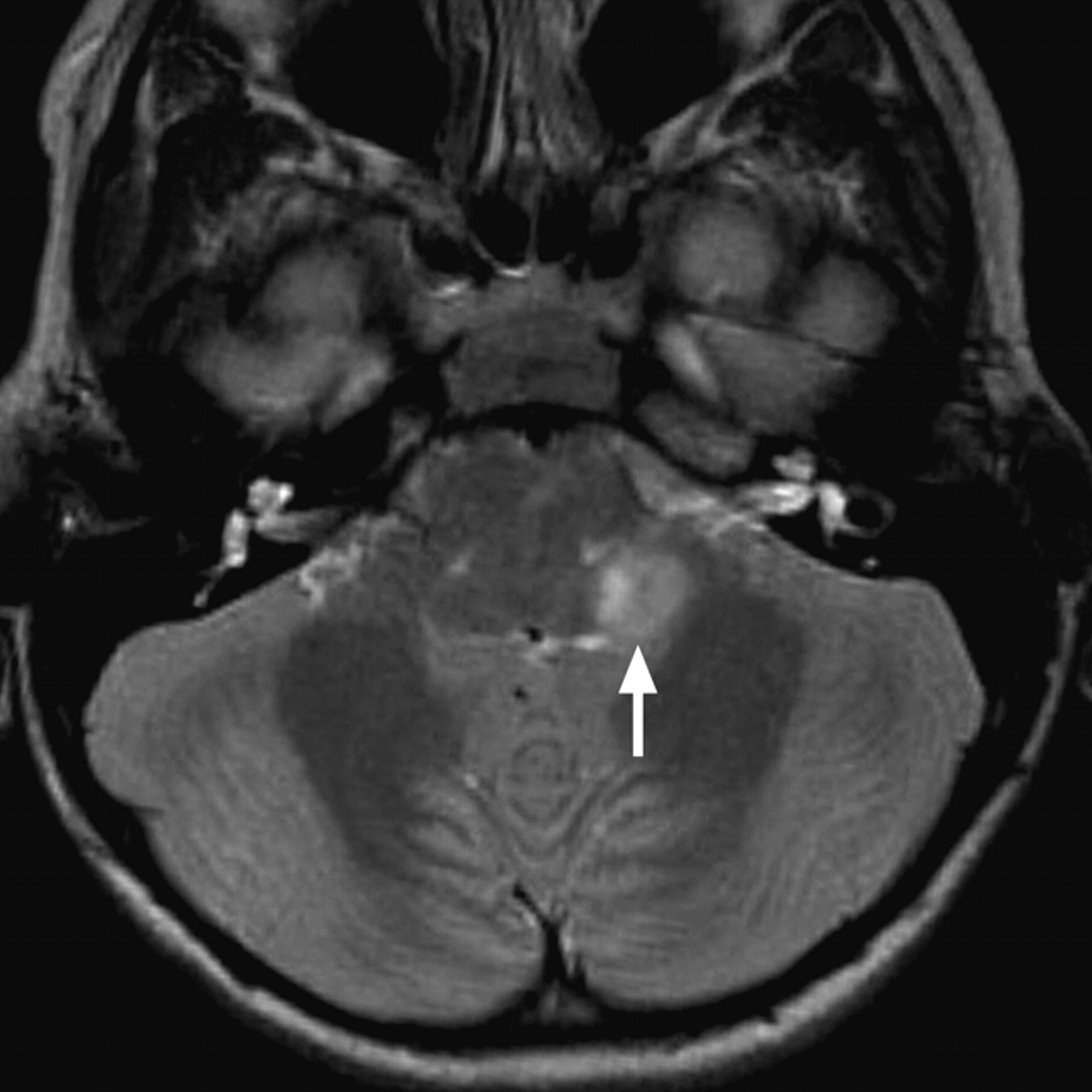
Tumors and other mass lesions of the IAC, cerebellopontine angle, and brain stem occasionally cause central SNHL in the children. Vestibular schwannomas can present with hearing loss, particularly in individuals with NF-2. Up to 18% of individuals with NF-2 present before 15 years of age, and hearing loss or tinnitus is the initial symptom in 20% of children who are ultimately diagnosed with the condition.45,46 The hearing loss in these cases is usually progressive and may be unilateral or bilateral. Characteristic brain imaging findings in NF-2 are the presence of multiple intracranial schwannomas (Fig 10 ) and meningiomas.

NF-2 axial gadolinium-enhanced T1WI through the level of the IACs demonstrates enhancing vestibular schwannomas in both IACs (arrows) and trigeminal schwannomas in both Meckel caves (arrowheads).
In addition, other posterior fossa masses, including cerebellar and brain stem pilocytic astrocytomas,47⇓–49 medulloblastomas,50 and cerebellopontine angle arachnoid cysts51,52 can also cause SNHL in children (Fig 11).

Pilocytic astrocytoma causing left-sided hearing loss. Axial T2WI demonstrates an exophytic hyperintense mass arising from the region of the left inferior cerebellar peduncle (arrow).
Conclusions
Although many of the syndromes associated with SNHL do not usually demonstrate gross inner ear anomalies by imaging, there are several in which inner ear malformations are common. In some cases, the presence of specific inner ear anomalies may be characteristic of certain syndromes—such as in BOR syndrome, Waardenburg syndrome, or X-linked deafness with stapes gusher—or may even be a defining feature of a syndrome, as in the case of CHARGE syndrome. SNHL presenting later in life is often related to inner ear infections or inflammatory conditions, trauma, or tumors—any of which may be suggested by findings at cross-sectional imaging. Neuroradiologists who routinely interpret temporal bone studies performed for the evaluation of childhood SNHL should be familiar with these potential causes and their respective imaging appearances; furthermore, they should be familiar with findings such as severe labyrinthine ossification, which might preclude or complicate cochlear implantation in a child with hearing loss.
Indicates open access to non-subscribers at www.ajnr.org
References
- © 2012 by American Journal of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Oval window perilymph fistula in child with recurrent meningitis and unilateral hearing loss
- A Case Series of X-Linked Deafness-2 with Sensorineural Hearing Loss, Stapes Fixation, and Perilymphatic Gusher: MR Imaging and Clinical Features of Hypothalamic Malformations
- The Unwound Cochlea: A Specific Imaging Marker of Branchio-Oto-Renal Syndrome
- Spectrum of Third Window Abnormalities: Semicircular Canal Dehiscence and Beyond
- Spectrum of Temporal Bone Abnormalities in Patients with Waardenburg Syndrome and SOX10 Mutations