
Abstract
SUMMARY: Brain imaging has progressed from exclusion of rare treatable mass lesions to a specific antemortem diagnosis. MR imaging−derived hippocampal atrophy and WMH are regarded as imaging biomarkers of AD and CVD respectively. Abnormal FP-CIT SPECT or cardiac iodobenzamide SPECT is a useful supportive imaging feature in the diagnosis of DLB. Frontal and/or anterior temporal atrophy and anterior defects on molecular imaging with FDG-PET or perfusion SPECT are characteristic of FTDs. Whole-body FDG-PET may be helpful in patients with rapidly progressing “autoimmune dementias,” and FLAIR and DWI are indicated in suspected CJD. A major role of imaging is in the development of new drugs and less costly biomarkers.
ABBREVIATIONS:
- apoE
- apolipoprotein E
- APP
- amyloid precursor protein
- bvFTD
- behavior variant frontotemporal dementia
- CJD
- Creutzfeldt-Jacob disease
- CVD
- cerebrovascular disease
- DLB
- dementia with Lewy bodies
- FP-CIT
- iodine-123-β-carbomethoxy-3 β-(4-iodophenyltropane) fluoropropyl
- FTD
- frontotemporal dementia
- MCI
- mild cognitive impairment
- PD
- Parkinson disease
- rCBF
- regional cerebral blood flow
- sCJD
- sporadic Creutzfeldt-Jacob disease
- TDP
- TAR deoxyribonucleic acid−binding protein 43
- VBM
- voxel-based morphometry
- vCJD
- variant Creutzfeldt-Jacob disease
- WMH
- white matter hyperintensities
The global prevalence of dementia is estimated to be 24 million and likely to double every 20 years.1 AD is the most common primary neurodegenerative disease but usually coexists with other pathologies associated with aging and dementia, most commonly cerebral small vessel disease.2 Less common but important causes of dementia are DLB and FTD. Primary dementing diseases have in common abnormal protein or peptide accumulation in the brain: τ and β amyloid in AD; α synuclein in DLB; and τ, TDP, or FUS (below) in FTD, as a result of either increased formation or decreased clearance.
North American and other guidelines recommend neuroimaging in patients with dementia.3 The indication has progressed from exclusion of a rare finding of treatable pathology to making a probable antemortem diagnosis, with the rationale that patients are better managed when the diagnosis is known. The diagnosis of a specific cause for dementia can still only be confirmed by brain biopsy or at postmortem, and imaging is not 100% specific. However, because of marked interindividual variability in the clinical manifestation of dementing illnesses, it can be argued, with some justification, that brain imaging gives a more specific representation of underlying neuropathology than clinical and psychometric assessment. This is particularly so for quantitative analysis of modern imaging modalities.4,5
One of the dilemmas of current diagnostic criteria is that once a patient meets those for a diagnosis of dementia, the disease responsible is extensive. Ideally a specific diagnosis would be made when the disease burden is too small to have caused significant impairment in activities of independent daily life, when treatment could arrest disease progression and prevent progressive decline and institutionalization. There are no such drugs on the market at present. However, brain imaging has an important role in drug development; once disease-modifying drugs are available, it may have a role in identifying the correct people to treat.
Alzheimer Disease
Background
In 2007 Dubois et al6 proposed a revision of the 1984 National Institute of Neurology Communicative Disorders and Stroke– Alzheimer Disease and Related Disorders Association criteria for AD to include not only a progressive episodic memory defect for at least 6 months but also ≥1 supportive neuroimaging or CSF biomarker. Imaging biomarkers include hippocampal atrophy on MR imaging or hypometabolism on FDG-PET (or other well-validated PET ligands). CSF biomarkers are increased total τ, increased phospho-τ, and decreased amyloid β1-42. The recently revised National Institute of Neurology Communicative Disorders and Stroke Alzheimer Disease and Related Disorders Association criteria7 include those for a diagnosis of dementia, probable AD, possible AD, and a final category with evidence of AD pathophysiology, similar to the criteria of Dubois et al.6 These revisions have placed imaging at center stage in both clinical practice and drug development.
The originally described pathologic features of AD are neurofibrillary tangles and extracellular β amyloid plaques.8 A brief overview of the current knowledge of the molecular pathology of AD is given to put imaging in AD in context.
Neuropathologically, τ pathology consists of abnormal hyperphosphorylated intracellular neurofibrillary tangles, which progress in characteristic stages from the entorhinal cortex to the neocortex9 with clinical evidence of dementia not being evident until neocortical stages 3–4. The stages of Braak and Braak9 correlate with cognitive impairment and result in neuronal dysfunction, synaptic loss, and cell death causing characteristic progressive atrophy beginning in the medial temporal lobes. Abnormal accumulation of β amyloid occurs in rare genetic forms of early-onset AD due to increased APP, abnormal APP processing (presenillin 1 and 2),10 or alterations in the β amyloid subunit of APP, resulting in cerebrovascular accumulation of β amyloid. The status of apoE is the major genetic influence on late-onset sporadic AD. The presence of apoE4 influences the earlier age of onset, greater cognitive impairment, and more rapid progression in AD. Increased amyloid formation and decreased clearance are thought to be the mechanisms by which apoE4 acts in AD, and it may also be associated with abnormal τ phosphorylation. However, being apoE4-positive is also associated with negative outcomes in other neurodegenerative diseases, such as PD,11 and it is possible that positive ε4 status is an indicator of a brain vulnerable to multiple neurodegenerative processes, not exclusively AD.
MR Imaging in Alzheimer Disease
While CT, particularly with the advent of helical CT, is a more readily available technique, MR imaging dominates the AD literature and allows more comprehensive brain assessment. Atrophy beginning in the entorhinal cortex and hippocampi is recognized as a structural imaging biomarker of AD.6,12 Hippocampal atrophy is a risk factor for cognitive decline and dementia in normal aging13 and for progressing from amnesic MCI to AD. The rate of hippocampal atrophy is more accurate than cross-sectional measurement,14 and a meta-analysis of rates of hippocampal atrophy found an annualized rate of 4.66% in patients with AD, compared with 1.41% in controls.15 Hippocampal and temporal gray matter atrophy are often found in cognitively healthy older people, but their presence is predictive of dementia and death in follow-up of well-characterized healthy cohorts.13,16
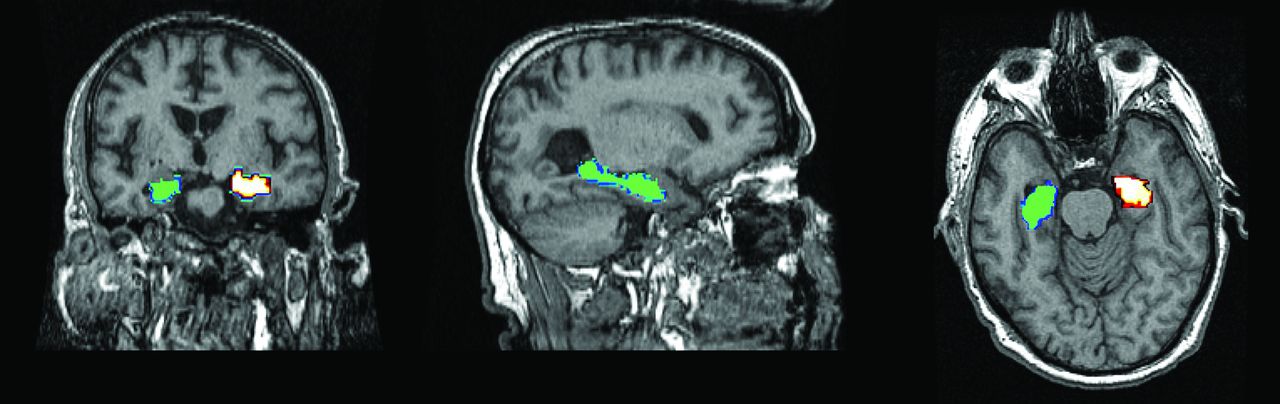
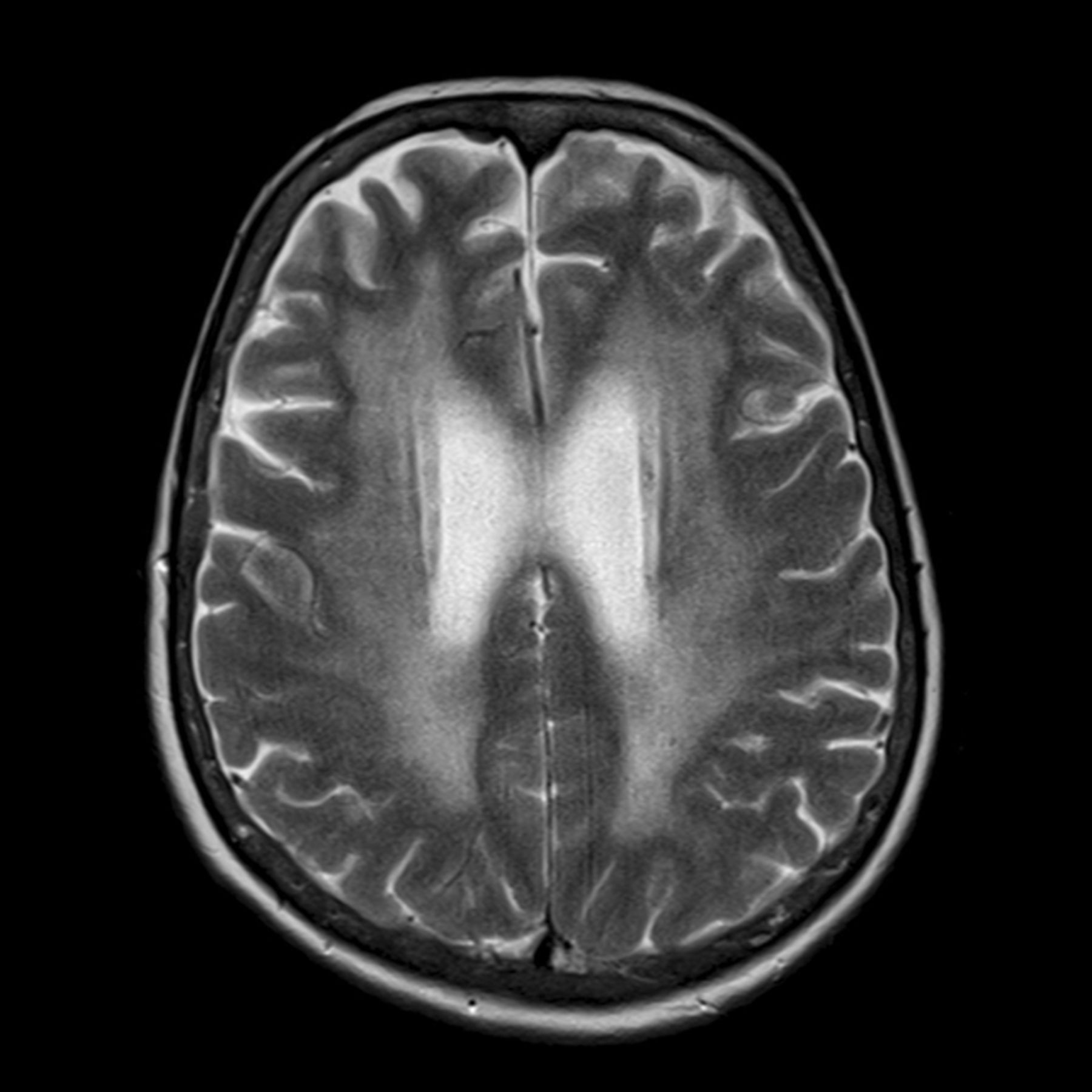
Methods of assessing hippocampal volume loss on structural MR imaging have progressed from visual assessment to manual hippocampal volumetry,17 with its inherent tedium and operator variability, to automated voxel-based methods. These include the Functional MR Imaging of the Brain Software Library (www.fmrib.ox.ac.uk/fsl) and FreeSurfer (surfer.nmr.mgh.harvard.edu), which are more suited to multicenter use and have better reproducibility than manual volumetry (Fig 1).

Automated voxel-based hippocampal volumetry by using FreeSurfer, showing left and right hippocampi.
Molecular Imaging with FGD-PET and Blood-Flow SPECT
Nonspecific molecular imaging with FDG-PET or rCBF SPECT have been documented for many years as useful imaging techniques in AD and in the prediction of which patients with amnesic MCI are at high risk of progression to AD.18 Both imaging modalities demonstrate characteristic deficits in posterior temporoparietal, posterior cingulate, and inferior frontal regions that reflect underlying neuronal dysfunction and neurodegeneration (Figs 2 A, -B and 3). The extent of reduced glucose uptake or blood flow has been shown to be greater than the result of atrophy alone and thus an earlier indicator of τ pathology than atrophy on structural imaging.19⇓–21 Quantitative analysis of FDG-PET and rCBF SPECT data by using voxel-based methods, such as statistical parametric mapping or 3D stereotactic surface projection, has improved image analysis and allows comparison of cases versus controls and disease progression in longitudinal studies.4 Such image-analysis methods are now available on most vendor-specific workstations and have improved the utility of molecular imaging in routine practice (Fig 2B). However, FDG-PET and rCBF SPECT demonstrate defects secondary to brain damage, and FDG and blood-flow ligands do not bind to AD pathology.
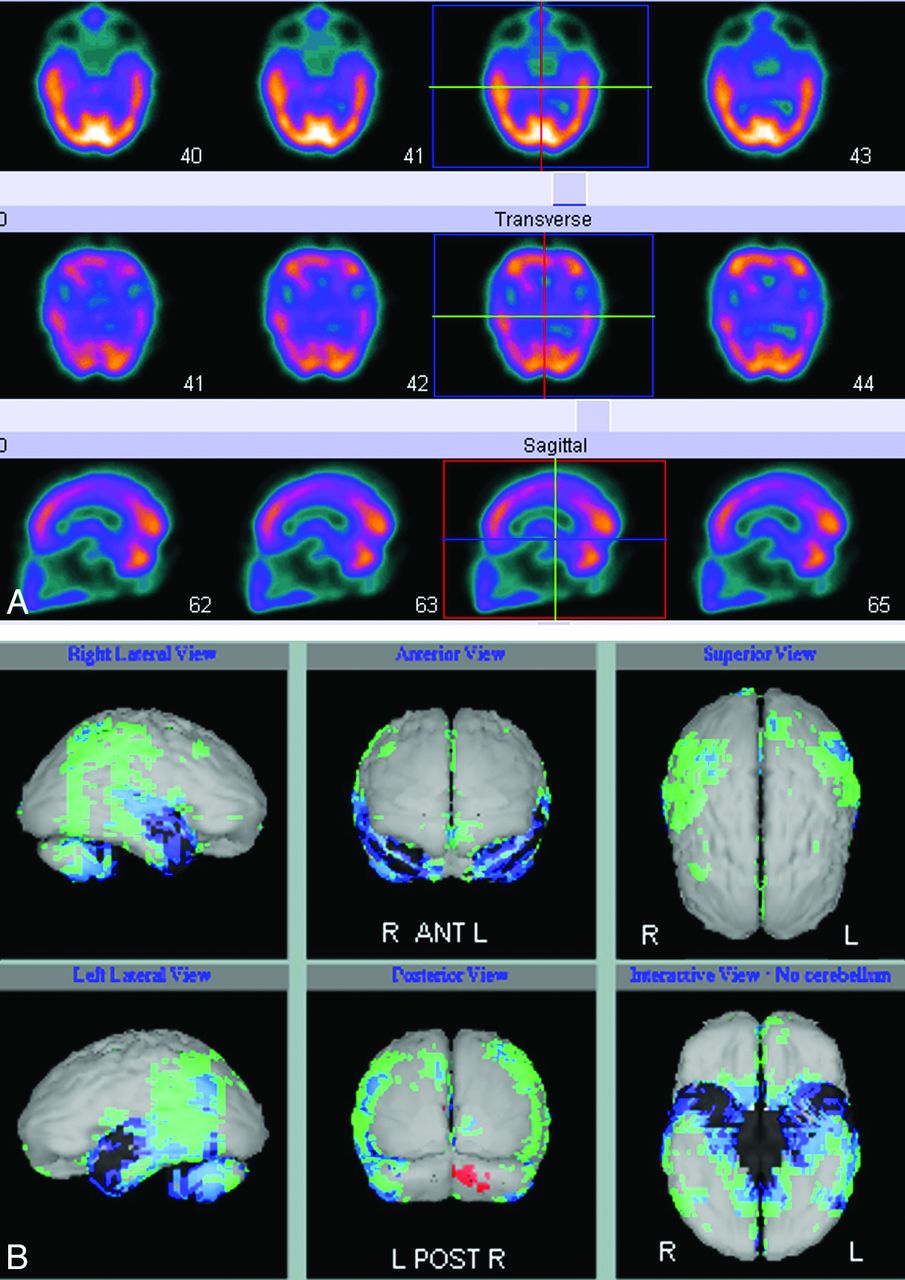
A, Blood flow (technetium TC99m hexamethylpropyleneamine oxime) SPECT showing reduced perfusion in the temporoparietal regions. B, Results of a comparison of A with a normal control data base highlights the temporoparietal deficits, in which perfusion is reduced between 2 and 3 SDs (blue and green regions).

18FDG-PET superimposed on T1-weighted MR imaging shows reduced FDG uptake in the medial temporal lobe (arrows) and corresponding atrophy (arrowheads) in early AD.
Molecular Imaging with Amyloid-Specific PET Ligands
The advent of specific β amyloid ligands, such as PIB, which is labeled with carbon 11,22 and flubetapir and flutemetamol, which are labeled with fluorine 18,23,24 brought great optimism for a new era in AD imaging diagnosis.
However, the degree of PIB uptake does not distinguish cognitively healthy older people, from those with amnesic MCI or from those with AD25; >20% of patients fulfilling research criteria for AD show negative PIB PET.26 Brain areas showing increased PIB uptake do not completely overlap typical sites of neurodegeneration; PIB binding does not increase with time27; not all patients with positive PIB PET develop AD; and the mechanism by which β amyloid leads to neurofibrillary tangle formation, synaptic loss, and cell death, characteristic of τ pathology is still not clear.28 In addition, in a comparison of early-onset (younger than 65 years) and late-onset (older than 65 years) AD, there was no difference in the extent or severity of PIB uptake, but metabolic defects on FDG-PET were, as expected, greater in the early-onset group.26 Positive PIB PET correlates with CSF amyloid β42, but there is poor correlation of PIB PET with hippocampal volume.29
While both τ (neurofibrillary tangles) and amyloid (plaques) have been regarded as hallmarks of AD, it is evident that these act on the brain, and, therefore, influence brain imaging at different phases of disease evolution. Jack et al21 have proposed a multiphase model of AD molecular pathology, which corresponds to established AD biomarker findings. The first phase is accumulation of β amyloid as extracellular plaques, and it is thought that the smallest β amyloid oligomers may be the most synaptotoxic form.
This is an early phase that can occur decades before cognitive decline and corresponds to reduction in CSF β amyloid and uptake of β amyloid markers on PET. The next phase is neurodegeneration due to intracellular accumulation of abnormal τ-paired helical filaments, which is associated with rising CSF τ (phospho-τ and total τ) and is first evident on imaging as reduced uptake in the temporal lobes and posterior cingulate on FDG-PET and blood-flow SPECT, due to neuronal dysfunction causing reduced glucose uptake and perfusion, respectively. This phase is followed by cell death, which corresponds to typical atrophy. This multiphase model is the best explanation we have at present for longitudinal changes in established biomarkers for AD. An alternative hypothesis is that β amyloid neuropathology and τ neuropathology are distinct processes that commonly coexist and probably have similar risk factors but that can occur independently. Possibly preexisting β amyloid pathology renders the brain much more vulnerable to the deleterious cognitive effects of τ neuropathology and thus the clinical expression of dementia.
The Alzheimer Disease Neuroimaging Institute (http://adni.loni.ucla.edu) collaboration is collecting a large dataset of imaging and other biomarkers of AD progression, including structural MR imaging, FDG-PET, PIB PET, CSF, and clinical findings. FDG-PET analyses have shown reduced FDG uptake in the posterior cingulate, precuneus, and parietotemporal and frontal cortices in those with probable AD and amnesic MCI.18 Longitudinal MR imaging analyses by using tensor-based morphometry have shown widespread atrophy in AD, with intermediate atrophy localized to the temporoparietal regions in MCI.30 Intuitively, combining MR imaging morphometry and CSF biomarkers improves the diagnostic classification of AD.31 The number of publications emerging from Alzheimer Disease Neuroimaging Institute and related initiatives in Europe and Australia is increasing exponentially and is an ideal opportunity for determining the future role of imaging in AD.
Cerebrovascular Disease
CVD is regarded as the most common secondary cause of dementia and is ubiquitous in the elderly, with most people older than of 60 years having some evidence of CVD on brain imaging. CVD can have differing appearances, depending on the site and size of the vessel involved, ranging from large cortical infarcts, lacunar infarcts, and macro- and microhemorrhage to white matter ischemia. These various imaging manifestations of CVD have been reported to have different associations with cognitive impairment and dementia, and a recent neuropathology series found leukoencephalopathy to be the main predictor of dementia.32
The terminology of white matter ischemia has varied; and “leukoencephalopathy,” “unidentified bright objects,” “cerebral small vessel disease,” “white matter lesions,” and “WMH” are synonyms used in the imaging literature. These are best demonstrated on MR imaging (Fig 4) and can be measured by using semiquantitative visual rating scales or quantitative voxel-based methods.33⇓–35 WMH are associated with vascular risk factors, particularly hypertension and impaired glycemic control.36 They are most common in the frontal white matter, increase linearly with age, and are associated with impaired fluid intelligence37,38 and executive dysfunction, features that characterize normal cognitive aging.39 WMH are also associated with adverse outcomes such as stroke, dementia, and death.40 It is now widely accepted that WMH are an imaging biomarker of vascular disease.41⇓–43

T2 axial MR imaging in a 68-year-old man with extensive WMH, typical of ischemic change.
Lacunar infarcts (or lacunes) occur in the subcortical white matter and basal ganglia and are also an imaging correlate of CVD.44 Perivascular spaces are pial-lined spaces around penetrating arteries and contain interstitial fluid. Enlarged perivascular spaces are frequently visualized on standard MR imaging brain examinations on older people and are more conspicuous at higher field strengths (Fig 5 A, -B). Enlarged perivascular spaces have been associated with vascular risk factors, such as hypertension, and are regarded as likely markers of cerebral small vessel disease, possibly due to increased vessel pulsatility.45

Enlarged perivascular spaces (A) on T2 axial MR imaging (arrow) and T1 sagittal MR imaging (B, arrow).
Cerebral hemorrhage is also evidence of CVD, with lobar macrohemorrhages having been traditionally associated with cerebral amyloid angiopathy. With increased use of MR imaging in population studies and particularly with the use of 3D T2* gradient-echo imaging, there has been increasing recognition of cerebral microhemorrhages or microbleeds in the healthy population.46 These increase with age and, in deep and infratentorial locations, are associated with vascular risk factors, WMH, and lacunar infarcts, while those in a lobar distribution are associated with apoE4 and diastolic hypertension (Fig 6).46,47
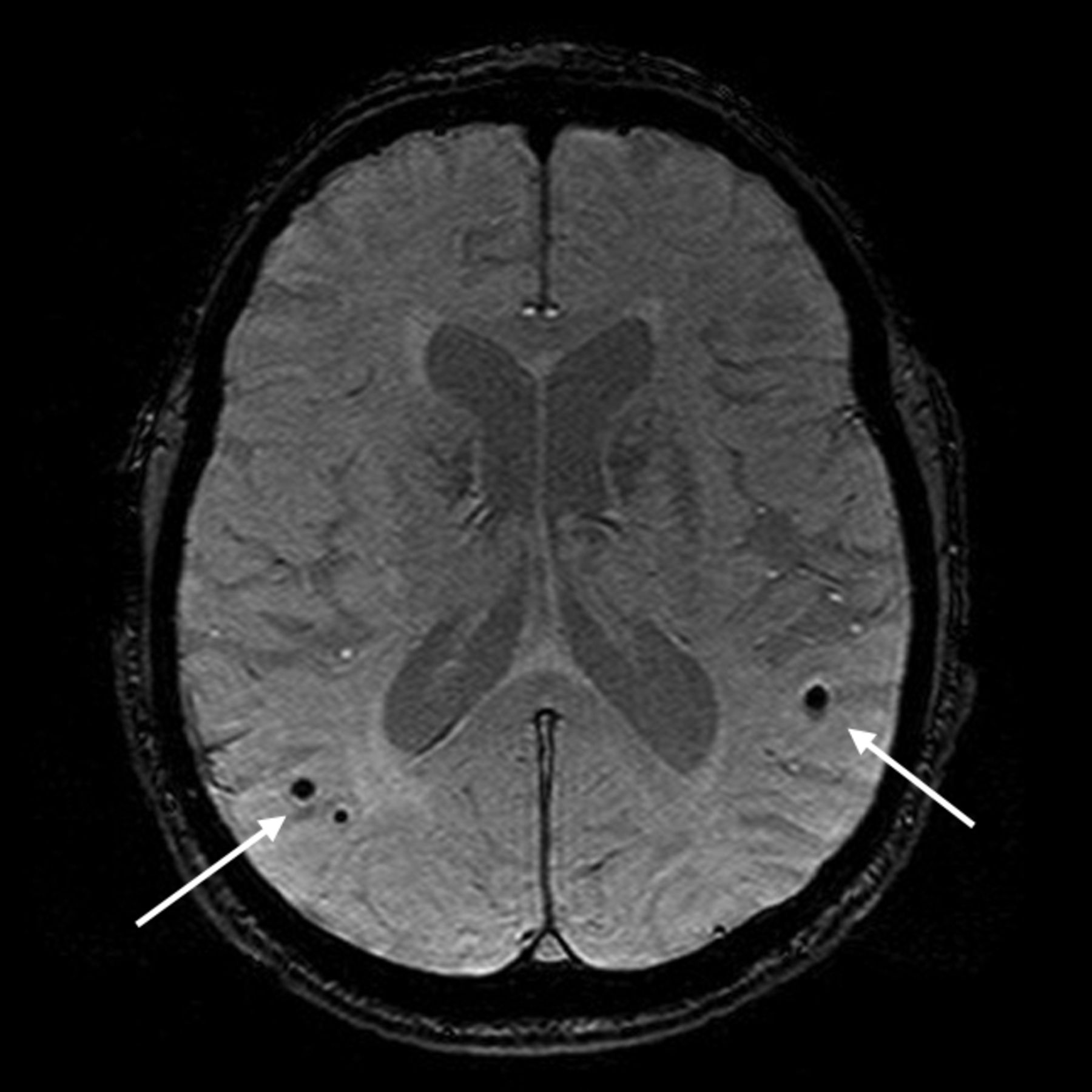
Lobar microhemorrhages on T2* gradient-recalled echo MR imaging (arrows). Images courtesy of Meike Vernooij, MD, Erasmus MC, Rotterdam, the Netherlands.
The presence of CVD on molecular imaging by using rCBF SPECT or FDG-PET characteristically shows defects in a patchy predominantly anterior distribution, and these correspond with WMH on MR imaging and impairments of executive function.48 The distribution is distinct from the posterior distribution seen in AD. However, on the basis of molecular imaging alone, it can occasionally be difficult to distinguish CVD from FTD, and structural correlation is helpful.
Frontotemporal Dementia
FTD is a heterogeneous group of diseases that account for approximately 5% of dementia and result in degeneration of the frontal and/or anterior temporal lobes and insula. Pathologically known as frontotemporal lobe degeneration, several different molecular pathologies and subtypes are recognized, which fall into 3 broad categories: 1) frontotemporal lobe degeneration-τ, which includes related tauopathies of progressive supranuclear palsy, corticobasal degeneration, and Pick disease; 2) frontotemporal lobe degeneration-TDP linked by abnormalities of the TDP, a subgoup of patients may also have motor neuron disease; and 3) frontotemporal lobe degeneration-fused in sarcoma, a group that have in common abnormal “fused in sarcoma” protein. The interested reader is referred to a recent consensus article.49
Clinically FTD is subdivided according to the predominant presenting feature into bvFTD, characterized by marked behavior and personality disorder, semantic dementia, and progressive nonfluent aphasia. Different FTD subtypes cause degeneration in specific brain regions and networks; for example bvFTD targets the anterior insula.50 These conditions tend to present earlier than AD and more often with a family history, but only a minority of patients meet diagnostic criteria at presentation.
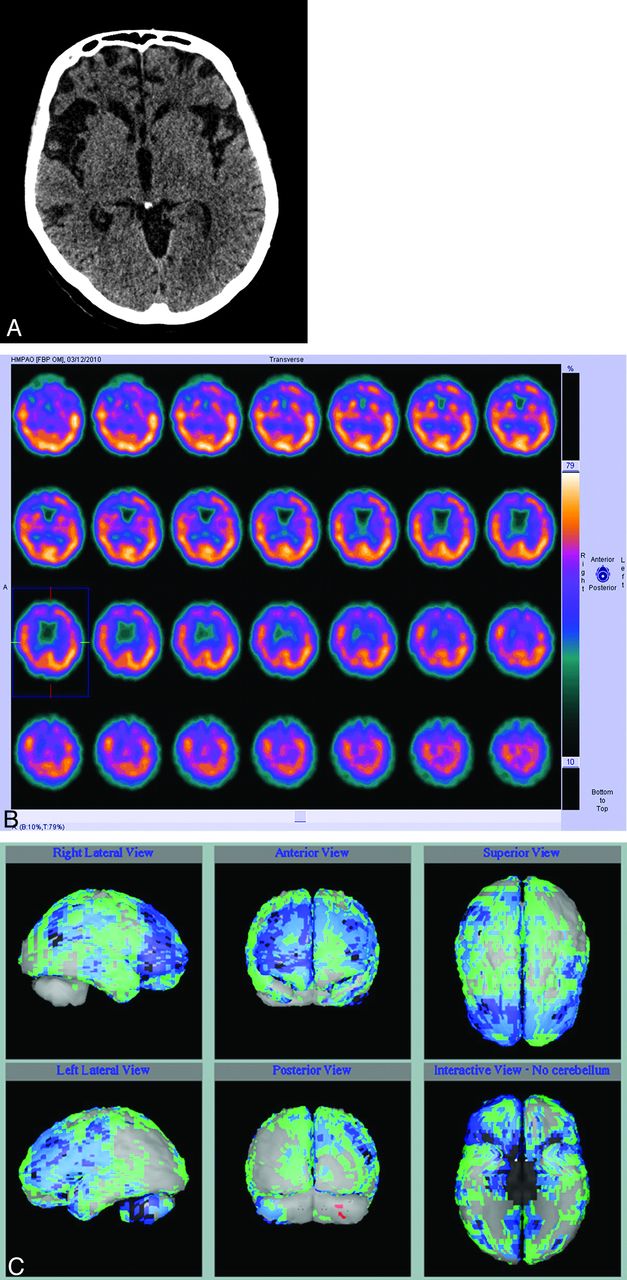
Until recent years, the best way of distinguishing FTD from AD in a patient with dementia in whom there was clinical doubt was molecular imaging with either FDG-PET or rCBF SPECT, both of which show anterior metabolic defects or perfusion defects, respectively, which are often asymmetric (Fig 7 A). Routine structural imaging with CT or MR imaging will show characteristic atrophy in the frontal lobes involving the ventromedial, orbitofrontal, anterior cingulate, anterior insula, and amygdala in established cases (Fig 7B). However, the most useful role of imaging is to suggest the diagnosis when the clinical picture is not clear, and both molecular imaging and quantitative analysis of 3D T1 MR imaging have good specificity in distinguishing FTD from AD based on identification of predominantly anterior-versus-posterior patterns of hypometabolism or atrophy.51,52 Pattern classification of VBM data in patients with dementia showed 100% accuracy in distinguishing patients with dementia from controls and 84% accuracy in separating FTD from AD.53
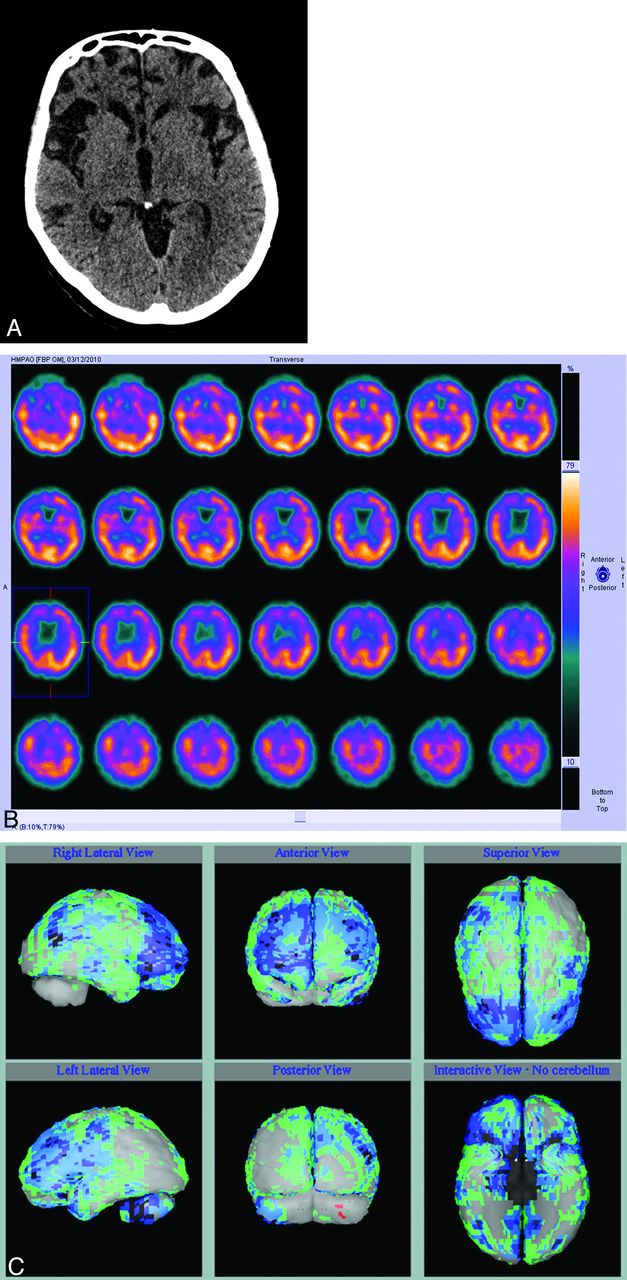
Axial CT image (A) in a patient with behavioral variant frontotemporal dementia shows a marked frontal atrophy, and axial perfusion SPECT images (B) show reduced anterior perfusion, which is more obviously appreciated in comparison with the normal control data base (C, as in Fig 2).
Dementia with Lewy Bodies
DLB accounts for approximately 15% of all dementia in the population and is clinically characterized by cognitive impairment with executive dysfunction, visuospatial impairment, visual hallucinations, motor parkinsonian features, and fluctuation in cognition and arousal. Rapid-eye-movement sleep-behavior disorder, severe neuroleptic sensitivity, and reduced striatal dopamine transporter activity on molecular neuroimaging are supportive features.54
There is evidence that the pathology of PD may begin in the peripheral sympathetic nervous system (below) and that the central nervous system is involved later. Pathologically DLB overlaps with PD and is characterized by dopaminergic cell loss and accumulation of α synuclein particles in presynaptic terminals that aggregate to form intracellular Lewy bodies. In a way similar to β amyloid pathology in AD, α synuclein can exist as oligomers, fibrils, and aggregates, and it is likely that the small oligomers are the most neurotoxic. These are predominantly found in the substantia nigra, pars compacta, and nigrostriatal projections in PD and in the cerebral cortex and limbic system in DLB, but widespread nigrostriatal degeneration is found in both and is present before clinical presentation.55 PD dementia is clinically and pathologically the same as DLB; the distinction is that in the former, motor symptoms predate cognitive decline by ≥12 months.54,56 However, increased β amyloid has also been found in DLB57 but not in PD dementia; and Pittsburgh Compound B PET showed cortical uptake in patients with DLB similar to that in patients with AD (the exception being the occipital lobes), which was significantly higher than cortical uptake patients with PD and healthy controls.58 This study also found some Pittsburgh Compound B uptake in more than half of the healthy control subjects.
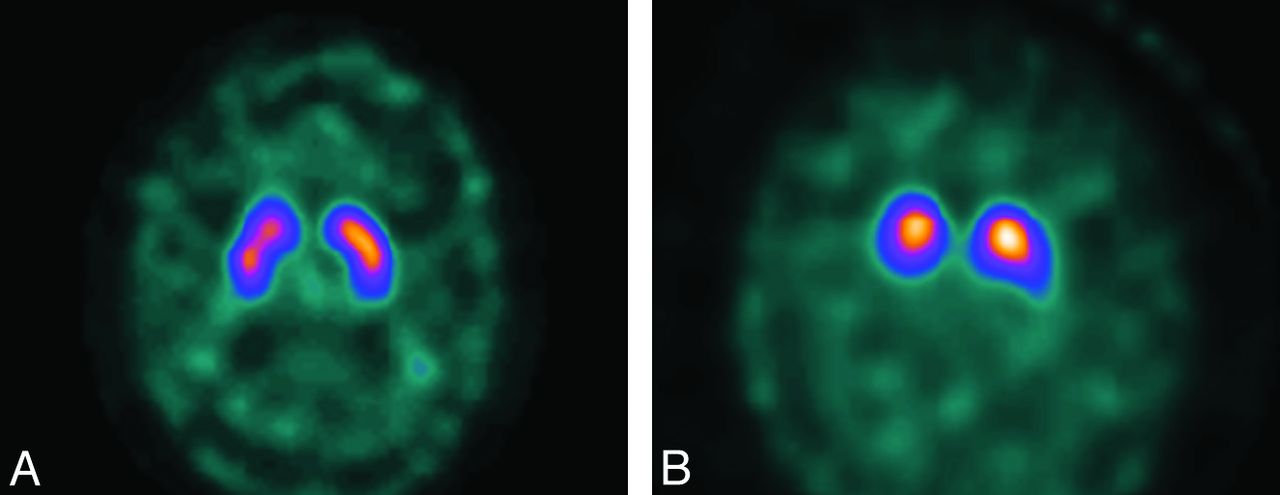
Diagnosis of DLB will often be evident clinically. However, it may be difficult in a substantial minority of patients. On FDG-PET and rCBF SPECT, the appearances of AD and DLB can be identical. Some reports have noted more widespread defects in DLB and involvement of the primary visual cortex, and differences between AD and DLB are more obvious with quantitative image analysis.59⇓–61 The introduction of dopamine transporter SPECT by using agents such as FP-CIT has made molecular imaging in PD and DLB more useful, with visual inspection of images sufficient to discern normal versus 3 grades of reduced uptake in the striatum (Fig 8 A, -B).62 Quantitative analysis of FP-CIT SPECT images, for example by using shape analysis, is an accurate and more reproducible method than visual assessment.63 Both fluorine-14 Dopa PET and FP-CIT SPECT have high positive predictive values in diagnosis of parkinsonian pathology, including DLB,64 and FP-CIT SPECT has been shown to be more accurate than clinical criteria in a longitudinal study of DLB with autopsy confirmation.65 However, findings of FP-CIT SPECT will inevitably be abnormal in patients with PD, making it inappropriate in patients with dementia with a pre-existing diagnosis of PD, who will, in most cases, have PD dementia or DLB.

FP-CIT images in a healthy patient (A) and a patient with parkinsonian disorder, in this case DLB (B).
There is much evidence, mainly in the Japanese literature, evaluating cardiac sympathetic imaging by using 18F Dopa PET and iodine 123 iodobenzamide in PD. This work indicates that reduced cardiac iodobenzamide (or F Dopa PET) uptake is an early finding in PD and DLB and that cardiac denervation precedes loss of neuronal cell bodies in paravertebral ganglia, suggesting a centripetal dieback process before central nervous system involvement.66
Structural MR imaging studies by using VBM have shown differences between DLB and AD, with greater atrophy in DLB in the striatum,61,67 midbrain, and hypothalamus, but the common finding is of relative preservation of the hippocampus in DLB compared with prominent atrophy in AD. MR imaging is also useful in Parkinson Plus syndromes, some of which cause dementia and are associated with specific structural MR imaging findings; for example, progressive supranuclear palsy results in characteristic atrophy of the midbrain giving rise to the “hummingbird” sign on sagittal images and the “Mickey Mouse” sign on axial images.68
Other Diseases Causing Dementia
While the diseases typically associated with dementia discussed above compose most dementia in clinical practice, it is important to be aware of the characteristic clinical and imaging findings of less common conditions that might present with dementia. The neuroradiologist is often the first to suggest such diagnoses and should be aware of the indications for specific imaging protocols, such as inclusion of DWI. Dementia has traditionally been divided into early and late onset with an arbitrary cutoff of 65 years of age. While most late-onset dementia will be AD, young-onset cases are usually due to the same conditions, namely AD, DLB, FTD, or cerebral small vessel disease, but are more likely to have a genetic association. However, it is rare for these diseases to present before the age of 35, when more unusual causes such as metabolic etiology should be considered.69
“Autoimmune dementia” has been proposed by researchers at the Mayo Clinic as a unifying term for a heterogeneous group of disorders that can cause dementia. These include various encephalopathies with specific clinical, electroencephalogram, or CSF features,70 previously referred to as “limbic encephalitis.” These may present with cognitive impairment, delirium, seizures; and most important, approximately 50% are responsive to steroids. Clinical features predicting a response to steroids are a subacute onset, fluctuating course, and tremor. Reported imaging findings are variable; MR imaging may show high signal intensity on T2-weighted images in involved brain areas. Molecular imaging with FDG-PET or rCBF SPECT may show foci of increased uptake or defects, and these imaging findings may resolve following response to steroids. Approximately half of patients with autoimmune dementia with neuron-specific CSF autoantibodies will have a paraneoplastic syndrome, and whole-body FDG-PET CT is appropriate to identify an underlying tumor.71
Rapidly progressive dementia is the typical clinical presentation of both sCJD and vCJD, and earlier age of onset is typical in vCJD.72,73 Other clinical features include hemiparesis, asymmetry, myoclonus in sCJD, painful sensory symptoms in vCJD, and characteristic electroencephalogram findings with periodic sharp-wave complexes.74 MR imaging in CJD shows typical high signal intensity on T2 and FLAIR in the pulvinar of the thalami in vCJD75 and in the caudate heads and cortex in sCJD, which can be asymmetric.74 These abnormalities are best demonstrated with DWI,76 which is crucial to include in patients with a rapidly progressive dementia; and the pulvinar sign is virtually pathognomonic of vCJD.
HIV-associated dementia is the most severe HIV-associated neurocognitive disorder, and the most common imaging manifestation of this is diffuse cerebral atrophy.77 Factor analysis of proton spectroscopic imaging data is effective in distinguishing those with HIV infection, characterized by an increased choline factor (consistent with glial proliferation), from those with HIV-associated dementia, characterized by a reduced N-acetylaspartate factor (consistent with neuronal cell dysfunction and death), and demonstrates the importance of white matter involvement in HIV.78
Conclusions
Modalities that hold promise for the future include fMRI, both task-related, in which relocation of task-related activation from the usual cortical regions may be one of the earliest findings of a failing brain,79 and intrinsic-connectivity network (resting-state) fMRI. Other MR imaging techniques, such as sodium MR imaging, can detect differences between a patient with mild AD and controls, which correlate with hippocampal volume.80 While these methods hold promise, unlike hippocampal atrophy on structural MR imaging, none are yet recognized or validated as an AD biomarker for the purposes of diagnosis or clinical trials.
One of the major current roles of imaging in dementia is in clinical trials of new drugs. Due to the relatively poor specificity of clinical assessment and because cognitive reserve81 will always confound the relationship between the severity of cognitive impairment and the extent of neuropathology, selection of patients for clinical trials based only on clinical criteria is inaccurate. Imaging is much closer to neuropathology and has a key role in clinical trials, both in selecting the population to treat, quantifying the extent of different neuropathologies, and quantifying treatment effect.82 Imaging in the context of dementia is likely to have the greatest use in quantifying the contribution from multiple neuropathologies so that these may be used in informative models of treatment response.
Currently available structural and molecular brain imaging modalities and modern image-analysis methods can produce beautiful images with a high predictive value for the underlying neuropathology. Unfortunately, lack of effective disease-modifying drugs means that imaging in dementia is not currently of health-economic benefit in routine clinical care. In the face of current global demographic change, it is also unlikely that we can afford to image every patient with dementia; hence, the need to develop cheap accessible biomarkers for dementing diseases. New developments in dementia include increased understanding of CSF biomarkers, though these are not yet at the stage of routine clinical diagnosis.83 Ultimately blood biomarkers would be of greatest clinical utility. The role of imaging will be in providing diagnostic confirmation during the development of such new diagnostic tests.
Footnotes
Disclosures: Alison D. Murray—CMES: Grants: work leading to publications referred to in a review on normal aging for which I am an author or coauthor was funded by grants from the Chief Scientist Office of the Scottish Executive, Alzheimer's Research Trust (now Alzheimer's Research UK), and the University of Aberdeen Development Trust. These funding bodies are acknowledged in the relevant publications;* Provision of Writing Assistance, Medicines, Equipment, or Administrative Support: Administrative and secretarial support in Aberdeen Biomedical Imaging Centre is funded by the University of Aberdeen and the Roland Sutton Academic Trust;* UNRELATED: Employment: main clinical academic contract, Comments: Funding for my post is provided to the University of Aberdeen by NHS Grampian;* Grants/Grants Pending: Current grants;* Payment for Lectures, Including Service on Speakers Bureaus: Eisai UK, Comments: £300 lecture fee to University of Aberdeen;* Patents: Wischik CM, Harbaran DV, Riedel G, Deiana S, Goatman EA, Wischik DJ, Murray AD, Staff RT. Case 25: Methods and materials for the use of diaminophenothiazines in the treatment of MCI. Patent/Published patent application 2008. GB 2008/002066 filed 19.06.2008;* Royalties: Springer, Comments: publication royalties from Practical Nuclear Medicine, 2nd ed, eds. Sharp, Gemmell, and Murray; Payment for Development of Educational Presentations: Eisai UK, Comments: £300 lecture fee to the University of Aberdeen;* Travel/Accommodations/Meeting Expenses Unrelated to Activities Listed: all travel expenses funded either from NHS study leave budget or University of Aberdeen; Other: Tau Therapeutics, Comments: spinout company of University of Aberdeen, which has conducted clinical trials in AD. I have carried out clinical reporting and image analysis for Phase II clinical trial as a member of staff of the university but receive no remuneration, either financial or in kind, for this activity.* (*Money paid to institution)
Indicates open access to non-subscribers at www.ajnr.org
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- © 2012 by American Journal of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.