
Abstract
SUMMARY: IgG4-related IPTs have been reported in various sites and may form part of the spectrum of systemic IgG4-related sclerosing disease. Some pseudotumors are clinically and radiologically indistinguishable from malignant tumors. We present the first case of an IgG4-related IPT of the trigeminal nerve diagnosed histopathologically without involvement of any of the common sites. The trigeminal nerve pseudotumor may represent a component of IgG4-related sclerosing disease.
Abbreviations
- ALK
- anaplastic lymphoma kinase
- DWI
- diffusion-weighted imaging
- HPF
- high-power field
- IgG
- immunoglobulin G
- IgG4
- immunoglobulin G4
- IMT
- Inflammatory myofibroblastic tumor
- IPT
- inflammatory pseudotumor
IPT is a rarely occurring non-neoplastic lesion that can mimic a neoplasm. Histologically, the lesions are characterized by inflammatory cell infiltration and variable fibrotic responses. IPT has been described in the literature by many different names, a fact that suggests the complexity and variable histologic characteristics and behavior of this entity.
In recent years, the relationship between some populations of IPT and IgG4 has been suggested. IgG4-related IPTs may form part of the spectrum of systemic IgG4-related sclerosing disease, with autoimmune pancreatitis being the predominant clinical presentation.1 However, IgG4-related sclerosing diseases without pancreatic involvement have also been reported.2
In this article, we report a rare case of pathologically proved IgG4-related IPT that involved the unilateral trigeminal nerve.
Case Report
A 59-year-old woman with a medical history of palmoplantar pustulosis and diabetes presented with left-sided facial numbness, which she had experienced for 4 years. She denied tinnitus, difficulty in hearing, or dysphagia. Physical examination revealed paresthesia in areas supplied by the left trigeminal nerve. Findings of the remainder of her neurologic examination were unremarkable. She had no surgical history in the head or neck region.
MR imaging revealed a homogeneously enhancing soft-tissue mass involving the skull base along the second and third divisions of the left trigeminal nerve (Fig 1A). T2-weighted imaging demonstrated a hypointense mass in the left Meckel cave, extending to the left pterygopalatine fossa via the left foramen rotundum and further to the infraorbital canal. The mass also extended to the left masticator space (Fig 1B) via the left foramen ovale. CT in a bone algorithm showed expansion of the left infraorbital canal (Fig 1C), the left foramen rotundum, and the left foramen ovale. There were no signs of bone destruction.
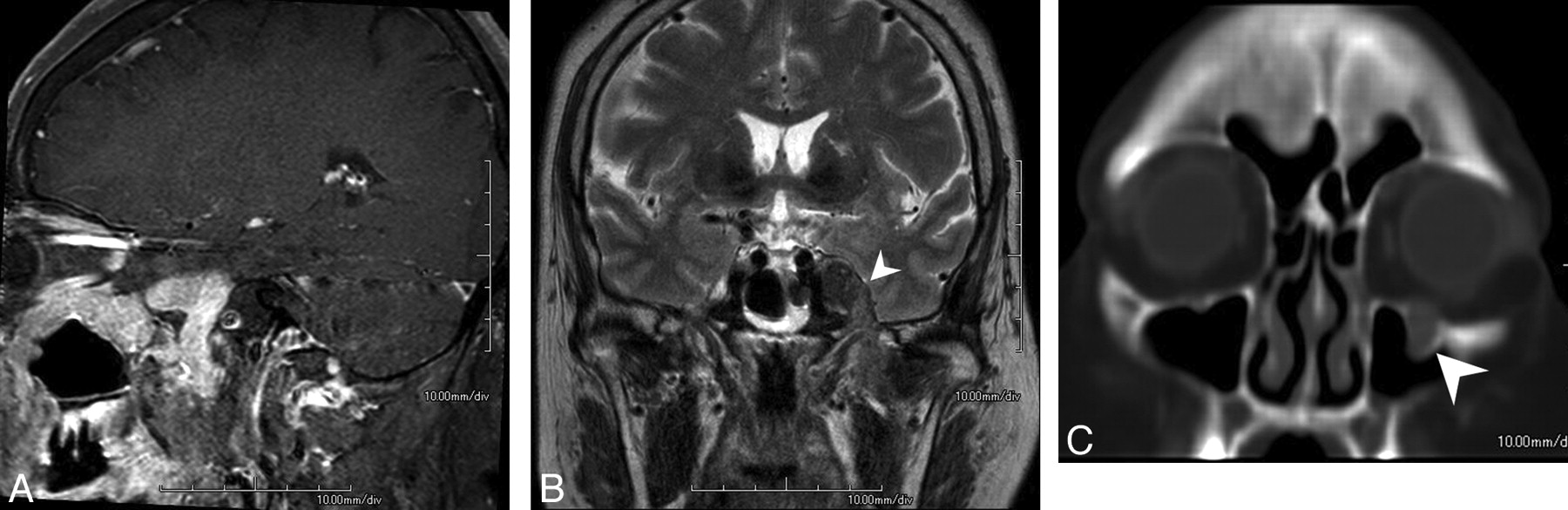
A, MR image reveals a homogeneously enhancing soft-tissue mass involving the skull base along the second and third divisions of the left trigeminal nerve. B, T2-weighted image demonstrates a hypointense mass (arrowhead) extending to the left masticator space via the left foramen ovale. C, CT scan in a bone algorithm shows enlargement of the left infraorbital canal (arrowhead).
Laboratory work-up results were all within normal limits. Tumor markers were also negative. Chest, abdominal, and pelvic CT findings were unremarkable.
The mass in the left retromaxillary area was surgically biopsied. Histologic evaluation showed nerve fibers (Fig 2A, arrowhead) surrounded by an attenuated inflammatory infiltrate (Fig 2B) comprising predominantly B and T lymphocytes with moderate fibrosis. Immunohistochemistry indicated abundant IgG4+ plasma cell infiltration (128/HPF) in and around the aggregates of lymphocytes (Fig 2C) and high ratios of IgG4+/IgG+ plasma cells (71%). There was no evidence of atypia or neoplasia. Immunostaining for ALK was negative. The final diagnosis was IgG4-related IPT.

A and B, Pathologic specimen in low power (A, magnification ×40) and high power (B, magnification ×200). Note nerve fibers (A, arrowhead) surrounded by a attenuated inflammatory infiltrate (B) with moderate fibrosis. C, (magnification × 200). Immunohistochemistry indicates abundant IgG4-positive plasma cell infiltration (128/HPF) in and around the aggregates of lymphocytes and high ratios of IgG4+/IgG+ plasma cells (>71%).
The patient did not seek further treatment. MR imaging studies at 6- and 12-month follow-up showed no significant changes.
Discussion
The etiology of IPT is still unclear but is thought to be infectious or autoimmune in nature. IPT is histologically characterized by a proliferation of spindle cells such as fibroblasts and myofibroblasts, admixed with inflammatory components consisting of lymphocytes, plasma cells, eosinophils, and histiocytes. However, the cell population can vary from one tumor to another or even from one microscopic field to another within the individual tumor.
Historically, IPT has been designated under various synonyms, such as plasma cell granuloma, xanthogranuloma, histiocytoma, and IMT. IMT is now considered as a “distinctive neoplastic proliferation” composed predominantly of myofibroblasts.3 Recent studies have revealed ALK gene rearrangements in IMT, suggesting a neoplastic cause, though, not all IMTs are positive for ALK by immunohistochemistry. Our case showed predominant lymphocyte proliferation with reactive fibrosis. Myofibroblastic spindle cells did not show cytologic atypia, and immunostaining for ALK was negative.
Recently, some populations of IPTs of the liver4 and lung5 have been reported to show pathologic characteristics similar to those of IgG4-related sclerosing disease as they involved lymphoplasmacytic infiltration and high IgG4+ plasma cell infiltrates. IgG4-related IPTs are occasionally complicated with IgG4-related pancreatitis and sialadenitis, suggesting that these types of IPTs may form part of the spectrum of systemic IgG4-related sclerosing disease.
There is currently no consensus on the number of IgG4+ plasma cells required for the diagnosis of IgG4-related IPT; however, several reports recommend that the ratio of IgG4+/IgG+ cells is also important for the diagnosis.4,5 According to the literature, IgG4+ cells >60–100/HPF and the ratio of IgG4+/IgG+ cells >40%-50% seem to be highly suggestive of IgG4-related disease. Yamamoto et al6 recently reported that both of these values are significantly lower in IMTs than in IgG4-related lesions. In our case, the number of IgG4+ plasma cells per HPF was 128, and the ratio of IgG4+/IgG+ cells was 71%.
In the head and neck region, IgG4-related IPTs of the salivary glands, lacrimal glands, and pituitary glands are well known; however, nasal/paranasal,7 dura mater,8 and parapharyngeal space9 lesions have also been reported. Our report describes a case of IgG4-related IPT of the trigeminal nerve without involvement of any of the preferential organs. Seol et al10 described another case of skull base IPT, which showed perineural spread along the trigeminal nerve, very similar to our case. However, no attempt was made to investigate the possible linkage with IgG4-related systemic disease or to characterize the plasma cell immunophenotype.
Imaging findings of IPT, including IgG4-related IPT, frequently reveal a homogeneously enhancing soft-tissue mass. On T2-weighted images, the mass is usually iso- to hypointense relative to brain. This can possibly be explained by the combination of fibrosis and attenuated cellularity of IPTs, which may also explain why IPTs tend to appear hyperintense on DWI. Attempts to differentiate IPTs from cancer11 and lymphoma12 on DWI have been made. However, the various degrees of cellularity, fibrosis, and perfusion in IPTs account for differences in diffusion restriction and often make the diagnosis difficult. IPTs with bone involvement and internal carotid artery encasement have also been reported in the past,13,14 including pathologically proved IgG4-related IPTs.7 In addition to these imaging features, our patient also had extensive perineural spread along the trigeminal nerve, highly suggestive of a neoplastic process.
IgG4-related IPT generally has a benign clinical course. These lesions usually respond to corticosteroids; however, relapse rates range from 30% to 40%.15 For unresponsive IPTs, surgical resection may be considered. Our patient did not seek further treatment. Her symptoms and follow-up MR images have not shown significant changes. However, a recent report suggests that there is a frequency of lymphomatous transformation in 10% of cases,16 adding yet another reason for close long-term surveillance.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received June 2, 2010.
- Accepted after revision June 20, 2010.
- © 2011 by American Journal of Neuroradiology
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A unique presentation of IgG4 disease with ocular, neurologic and mastoid involvement
- Implications of enlarged infraorbital nerve in idiopathic orbital inflammatory disease
- Intracranial spread of IgG4-related disease via skull base foramina
- Immunoglobulin G4-Related Disease of the Orbit: Imaging Features in 27 Patients
- Ocular adnexal IgG4-related disease: CT and MRI findings
- MR Imaging of IgG4-Related Disease in the Head and Neck and Brain
- Bilateral inflammatory pseudotumour of the trigeminal nerve: a diagnostic challenge