
Abstract
BACKGROUND AND PURPOSE: CLD is a rapidly progressive and invariably fatal neurodegenerative disorder. We describe clinical and neuroimaging findings in 5 infants with CLD.
MATERIALS AND METHODS: Retrospective review of medical records of infants with CLD from the past 11 years at our institution was performed. Relevant clinical and demographic data were recorded. Specific attention was directed toward postmortem examination findings and genetic testing. CT and MR imaging results were reviewed.
RESULTS: Five Cree infants were diagnosed with CLD. CT demonstrated bilateral symmetric hypoattenuation of the white matter and globus pallidus. MR imaging demonstrated corresponding T2 hyperintensity in these regions and abnormal signal intensity in the thalami and substantia nigra. Symmetric restricted diffusion in the deep white matter was seen. MRS demonstrated decreased NAA, elevated choline, and the presence of lactate. Postmortem examination in 1 infant showed corresponding poor myelination in the brain stem, cerebellum, deep gray structures, and the cerebral hemispheres. Genetic testing in 2 infants revealed homozygous mutations in the eIF2B5 gene.
CONCLUSIONS: Neuroimaging in CLD is striking and is an important tool in diagnosing CLD. Extensive white matter involvement as well as involvement of the globus pallidus and patchy involvement of the thalami and substantia nigra are characteristic. MRS findings are compatible with destruction of normal brain parenchyma with evidence of anaerobic metabolism in the regions of demyelination. Clinical suspicion of VWM in a Native American infant from this region should prompt the consideration of CLD with appropriate imaging work-up and genetic testing.
Abbreviations
- ADC
- apparent diffusion coefficient
- CACH
- childhood ataxia with central hypomyelination syndrome
- Cho
- choline
- CLD
- Cree leukodystrophy
- Cr
- creatine
- DNA
- deoxyribonucleic acid
- DWI
- diffusion-weighted imaging
- eIF2
- eukaryotic translation initiation factor
- eIF2B
- eukaryotic initiation factor 2B
- eIF2B5
- eukaryotic initiation factor 2B subunit 5
- FLAIR
- fluid-attenuated inversion recovery
- IV
- intravenous
- Lac
- lactate
- MRI
- MR imaging
- MRS
- MR spectroscopy
- NAA
- N-acetylaspartate
- PRESS
- point-resolved spectroscopic sequence
- RNA
- ribonucleic acid
- VWM
- vanishing white matter disease
CLD is an autosomal recessive neurologic disorder that affects infants of Cree and Chippewayan descent. It was previously thought to occur exclusively in northern Quebec and Manitoba, but a single case has been reported in the Western Canadian province of Saskatchewan.1,2 The onset is typically between 3 and 9 months of age and affects previously healthy infants, usually following a viral infection or trauma.3
Previous studies have demonstrated that the imaging characteristics of CLD are similar to those seen in patients with CACH, also known as VWM.2 Recently, a genetic basis for CLD has found it allelic to VWM. CLD is, therefore, now considered a severe variant of VWM/CACH.3 Mutations in eIF2B are found in patients with CACH/VWM and ovarian leukodystrophy.4
We describe the clinical and radiographic findings in a series of 5 infants with CLD from the Western Canadian province of Saskatchewan.
Materials and Methods
The medical records of infants diagnosed with CLD at the University of Saskatchewan from March 1996 to September 2005 were reviewed. This study was approved through the institutional Biomedical Research Ethics Board. Data including age, sex, clinical presentation, and family history were obtained. Neuroimaging (CT, MR imaging, and MRS), pathology, and genetic studies were reviewed.
Incremental unenhanced 2.5-mm axial CT images were acquired from the skull base to the vertex. MR imaging and MRS were performed on a 1.5T whole-body imaging system by using a standard quadrature head coil (Magnetom SP before 1996 and Symphony after 1996; Siemens, Erlangen, Germany) with a 25-mT/m gradient (slew rate, 120 T/m/s). Some or all of the following acquisition parameters were used on each infant by using an FOV of 23 cm: T1-weighted imaging (TR = 400–609 ms, TE = 12–15 ms), T2-weighted imaging (TR = 3990–4010 ms, TE = 96–121 ms), FLAIR (TR = 9000 ms, TE = 71–73 ms), and DWI (TR = 109 ms, TE = 3600 ms). Diffusion-weighted 3-scan trace in the transverse plane by using echo-planar imaging was performed with b-values of b=0, b=500, and b=1000, acquisition time 1 minute 18 seconds. ADC maps were derived automatically on a voxel-by-voxel basis by using commercially available software. For contrast-enhanced imaging, 2 mL of IV gadolinium (0.2 mL/Kg) contrast agent was administered. MRS was performed by using a multivoxel PRESS acquisition technique with the following parameters: section thickness = 15 mm, TR = 3000 ms, TE = 20 ms, NEX = 4, and 8 phase-encoding steps. All images were subjectively evaluated by the same staff neuroradiologist (S.H.). Images were scrutinized for the pattern/extent of involvement. Where possible, cases were assessed for diffusion restriction and abnormal metabolic profile.
Infants 4 and 5 underwent genetic testing for known eIF2B mutations. Infant 2 underwent a postmortem examination with gross and microscopic examination of the brain. Following fixation of the brain in 10% formalin, representative tissue blocks were procured from the brain stem, cerebellum, deep gray structures, and cerebral hemispheres. These were subsequently embedded in paraffin and cut into 5-μm-thick sections. Staining was performed with hematoxylin-eosin, Luxol fast blue stain for myelin, and Bielschowsky silver stain for axons.
Results
Clinical and Demographic Findings
Five (4 female, 1 male) previously healthy Cree infants ranging in age from 5 to 12 months (median age, 7 months) were identified. All infants were of Cree descent and resided in Saskatchewan. All infants presented with a rapidly progressive neurodegenerative course following a mild upper respiratory tract infection. The average age of onset was 8 months. Clinical features consisted of seizures and spasticity with progression to death within 6 months of presentation. Infants 3 and 5 were siblings. The clinical details are summarized in Table 1.
Clinical details of presented cases
Neuroimaging Findings
CT.
In the 4 infants with CT imaging (Table 1), there was bilateral and symmetric diffuse hypoattenuation of the cerebral and cerebellar white matter, internal capsule, corpus callosum, globus pallidus, and brain stem (Fig 1A–C). The thalamus was only involved in infant 3. There was notable sparing of the caudate nuclei, pons, and putamen in all infants. CT scans in infant 1 had been destroyed and could not be included. Contrast-enhanced CT was not performed.

Infant 2, an 11-month-old Cree girl with hypotonia and developmental regression. A−C, Axial CT images demonstrate diffuse bilateral symmetric hypoattenuation of the cerebral and cerebellar white matter. D−F, Axial T2 images demonstrate bilateral symmetric T2 hyperintensity of the cerebral white matter, substantia nigra (small white arrow), and globus pallidus (large white arrow), with involvement of the thalami (black arrow) and sparing of the caudate nuclei, red nucleus, and putamen.
MR Imaging.
All 5 infants underwent MR imaging (Table 2). T2-weighted images (Fig 1D–F) demonstrated hyperintensity in the cerebral subcortical white matter, external capsule, extreme capsule, corpus callosum, corticospinal tracts, cerebellum, globus pallidus, midbrain, and pons with patchy involvement of the thalami and substantia nigra. There was occasional patchy involvement of the caudate nuclei (1 infant) and putamen (1 infant). The red nucleus and claustrum were spared in all infants. In all infants who had FLAIR sequences (infants 3, 4, and 5), there were linear and punctate regions or “islands” of increased signal intensity on the FLAIR sequences (Fig 2). In these infants, most of the remaining cerebral white matter was hypointense on the FLAIR sequences (with signal intensity approaching that of CSF). Only infant 4 received contrast, but no abnormal enhancement was visualized. In patients who underwent DWI (infants 4 and 5), an unusual pattern was seen with symmetric regions of increased diffusion in the white matter, particularly in the frontal regions (Fig 3 ). There were also symmetric regions of diffusion restriction involving the corpus callosum and the subcortical U-fibers in the parieto-occipital region. Details of variably involved structures can be seen in Table 2.
Variably hyperintense brain structures on T2-weighted MRI of 5 infants with known CLD

Infant 5, an 8-month-old Cree girl with hypotonia and developmental regression. Axial FLAIR image demonstrates diffusely hypointense white matter with scattered linear and punctate foci of increased signal intensity.

Infant 4, an 8-month-old Cree boy with lethargy, hypotonia, and seizures. A and B, Axial b=1000 (A) and ADC maps (B) demonstrate regions of diffusion restriction in the corpus callosum (white arrows) and posterior subcortical U-fibers (small white arrows). C and D, T2 (C) and FLAIR (D) images are provided for comparison.
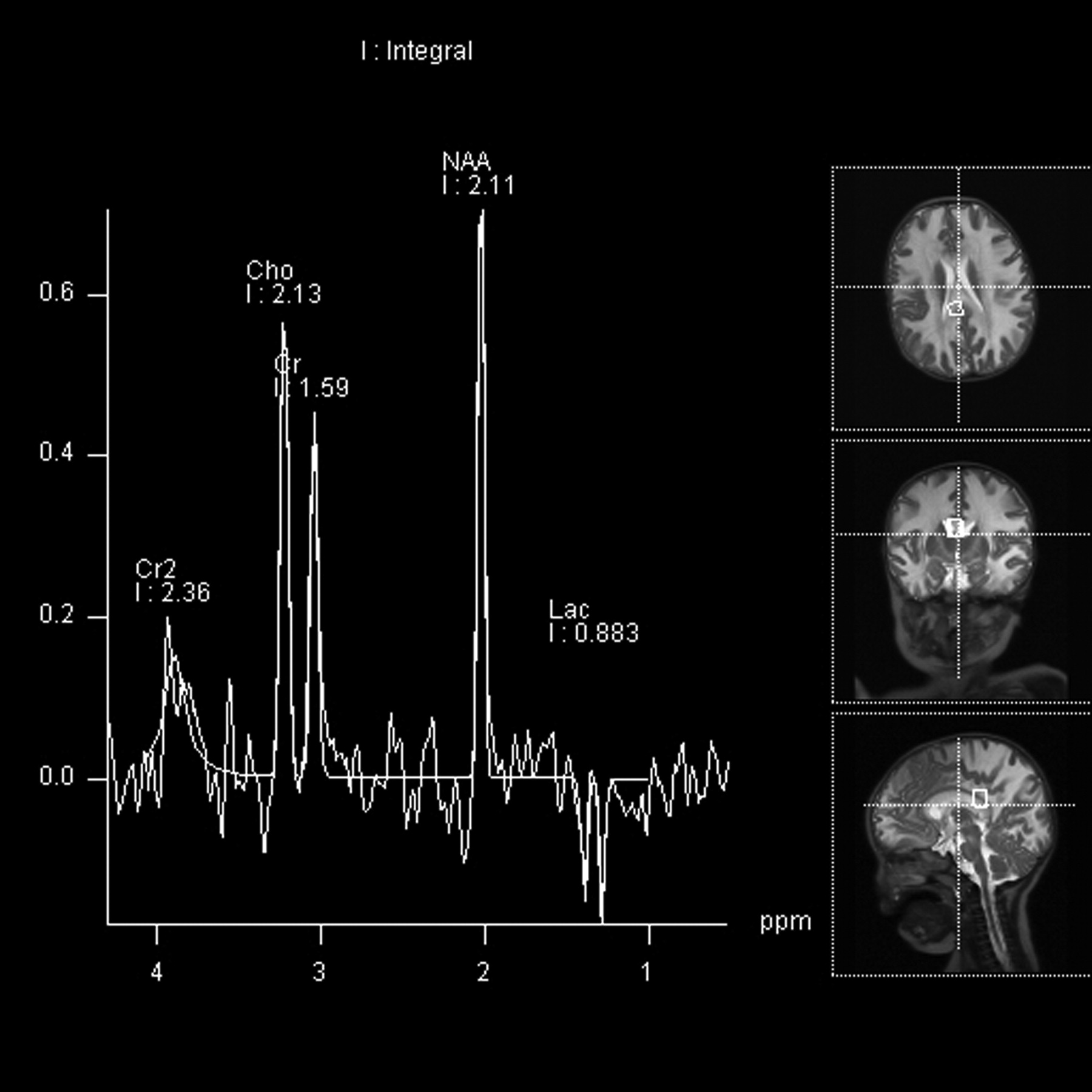
MRS.
Two infants underwent MRS. The multivoxel PRESS acquisition (Fig 4) demonstrated diminished NAA in keeping with neuronal loss or dysfunction, elevated choline-to-creatine ratio in keeping with demyelination, and the presence of lactate in keeping with anaerobic metabolism in the affected white matter.

Infant 4, an 8-month-old Cree boy with lethargy, hypotonia, and seizures. Multivoxel PRESS acquisitions (TR/TE = 3000/144 ms). A voxel in the region of the splenium of the corpus callosum demonstrates diminished NAA, minimally elevated choline, and a lactate peak.
Genetic and Pathologic Findings
DNA Analysis.
The 2 infants who underwent DNA testing were homozygous for the previously described G594A mutation in the eIF2B5 gene known to cause CLD.
Pathologic Findings.
Only infant 2 underwent postmortem examination. Examination of non-central nervous system organs revealed an acute bronchopneumonia only. The brain weighed 950 g before fixation, without any external abnormalities evident. Cut sections of the brain revealed somewhat soft grayish-white matter of the cerebral hemispheres, including the deep structures. These gross abnormalities were less prominent in the brain stem and cerebellum. Microscopically, white matter from all sites examined (including the medulla, pons, midbrain, cerebellar hemisphere, deep gray structures, hippocampus, deep cerebral white matter, and various neocortical sites) showed variable myelin pallor with Luxol fast blue staining, microvacuolation/rarefaction, and associated astrocytic gliosis. There were occasional foamy macrophages with little evidence of active myelin breakdown. These changes were most prominent in deep cerebellar and cerebral hemispheric white matter, as well as in the internal capsule and corpus callosum, where microvacuolation/rarefaction was approaching early cavitation. Subcortical arcuate fibers, subependymal white matter, and white matter of cerebellar folia showed mild preservation. Gray matter structures throughout the brain appeared preserved. Intracellular storage products and inflammation were not present. These neuropathologic findings are similar to those reported in patients with VWM/CACH by Dietrich et al.5
Discussion
CLD is a demyelinating disease that is known to affect infants of Cree and Chippewayan descent. Previously, the diagnosis was based on the clinical presentation and neuroimaging findings. Recently, CLD has been linked to homozygous missense mutations in the eIF2B5 gene, resulting in histidine replacing arginine at position 195 of the ε subunit of eIF2B. eIF2B converts eIF2 (the protein synthesis initiation factor) from an inactive form to an active form, which plays an essential role in the initiation of messenger RNA translation.4,6 It also regulates the translation of RNA to protein, particularly under stress conditions.7 VWM, a less severe but progressive neurologic disorder seen predominantly in older children of NorthernEuropean ancestry,3 has been associated with mutations in any of the eIF2B5 subunit genes (eIF2B1–5).8 This not only supports the molecular basis of CLD but also demonstrates that CLD is allelic to VWM.
Two theories have been postulated as to how this missense mutation leads to the diffuse demyelination seen in CLD. The first hypothesis is that this mutation causes the accumulation of denatured proteins, whereas the second theory postulates that this missense mutation leads to a lack of eIF2 recycling and subsequent decreased rates of protein translation initiation and cell growth.9,10 The precise reason for the susceptibility of cerebral white matter to eIF2B dysfunction has yet to be determined. This recent advance in the molecular basis of the disease has allowed the genetic confirmation of the diagnosis and the recognition of carriers for the disease.4
CLD affects developmentally healthy infants between the ages of 3 and 9 months. Although our case series includes 4 female infants and 1 male infant, a sex predilection, to our knowledge, has not been described in the reviewed literature. The disease is often preceded by an upper respiratory tract illness or minor trauma and is followed by seizures, spasticity, decreased level of consciousness, and eventual progression to death.1 CLD is invariably fatal by 21 months of age.
The neuroimaging hallmark of CLD has been said to be diffuse extensive symmetric white matter abnormality in the brain stem, cerebellum, and cerebrum with sparing of the caudate nuclei and putamen on MR imaging.2 The infants in our review had neuroimaging findings similar to those previously described in the literature. There were changes affecting the periventricular, lobar, and subcortical white matter identified early after the onset of illness. CT demonstrated diffuse symmetric hypoattenuation of the cerebral and cerebellar white matter, globus pallidus, and brain stem with sparing of the caudate nuclei and putamen. MR imaging demonstrated corresponding T2 hyperintensity in these regions with sparing of the red nucleus. Patchy involvement of the thalami, caudate nuclei, putamen, and substantia nigra was noted in our series.
Based on the neuroimaging findings alone, there are differential diagnostic considerations for this appearance, including leukodystrophies that predominantly involve the white matter, which include Canavan, Alexander, and VWM diseases. The former 2 classically present with macrocephaly, which is an important distinguishing finding when present. In addition, Canavan disease presents in a different ethnic group, and Alexander disease tends to classically demonstrate periventricular frontal enhancement after contrast administration. Macrocephaly and contrast enhancement have not been described with CLD. Canavan disease has a similar appearance, but the pattern of white matter involvement is usually centripetal, beginning in the subcortical arcuate fibers.2 Alexander disease is also similar, but it is usually predominantly frontal in distribution with cysts and ventricular enlargement. Severe acute disseminated encephalomyelitis could also be considered, but this entity almost always presents in older children and tends to be less symmetric, often with associated enhancement and mass effect.
VWM/CACH has a similar appearance, as expected, because CLD is considered within this spectrum of disease. A categorization system described by van der Knaap et al10 suggests that CLD would be a category B2 leukoencephalopathy, similar to VWM. The most important distinction is clinical: VWM has a slower clinical course and a later onset.2 Involvement of deep gray matter in CLD is another distinguishing feature. While the neuroimaging features may also be in keeping with other leukodystrophies, rapid progression along with Cree or Chippewayan ethnicity is highly suggestive of CLD.
Clinically, CLD may behave like Krabbe disease; however, the difference can be discerned on neuroimaging. Krabbe disease may have hyperattenuations in the thalami, corona radiata, and cerebellar cortex on CT and high T2 signal intensity in the periventricular and cerebellar white matter on MR imaging. Krabbe disease also has generally progressive brain atrophy.11 In contrast, CLD diffusely affects the white matter with no preservation of the subcortical region and little or no atrophy.
The findings noted on DWI showing regional differences with increased diffusibility anteriorly and diffusion restriction related to the corpus callosum and in the subcortical U-fibers of the parieto-occipital lobes have not been previously described in VWM, to our knowledge. Although no enhancement was seen, this may represent an acute demyelinating margin, similar to that seen in adrenoleukodystrophy.
MRS demonstrated diminished NAA in affected white matter, in keeping with neuronal loss or dysfunction. Elevated choline was also seen and has been described in the setting of demyelination. The presence of lactate is in keeping with anaerobic metabolism. These features suggest destruction of normal brain parenchyma as well as metabolic stress in the regions of demyelination.
This retrospective study has some limitations. Most notably, genetic testing and postmortem examination were not performed on all of the infants in our study. Genetic testing only became available for the last 2 infants included in this series. Retrospective tissue analysis was attempted on previous samples but was not successful.
Conclusions
CLD is a rapidly progressive neurodegenerative disease that affects Native American infants of Cree and Chippewayan descent. Our series of infants are the first reported cases of CLD in Western Canada. Clinical suspicion of VWM in a Native American infant from this region should prompt the consideration of CLD with appropriate imaging and genetic testing.
Footnotes
-
Paper previously presented in part at: Annual Meeting of the American Society of Neuroradiology, April 29–May 5, 2006; San Diego, California.
References
- Received June 25, 2008.
- Accepted after revision March 4, 2010.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.