
Abstract
SUMMARY: The purpose of this article was to review the embryologic stages of the cerebral cortex, illustrate the classification of disorders of cortical formation, and finally describe the main MR imaging features of these disorders. Disorders of cortical formation are classified according to the embryologic stage of the cerebral cortex at which the abnormality occurred. MR imaging shows diminished cortical thickness and sulcation in microcephaly, enlarged dysplastic cortex in hemimegalencephaly, and ipsilateral focal cortical thickening with radial hyperintense bands in focal cortical dysplasia. MR imaging detects smooth brain in classic lissencephaly, the nodular cortex with cobblestone cortex with congenital muscular dystrophy, and the ectopic position of the gray matter with heterotopias. MR imaging can detect polymicrogyria and related syndromes as well as the types of schizencephaly. We concluded that MR imaging is essential to demonstrate the morphology, distribution, and extent of different disorders of cortical formation as well as the associated anomalies and related syndromes.
Disorders of cortical formation are a heterogeneous group characterized by an abnormal structure of the cerebral cortex. They are caused by interruption of the normal developmental sequences due to a lack of normal gene expression, by production of an abnormal gene, or by interruption of the function of a gene by external causes such as infection or ischemia. There are different classifications of the disorders of cortical formation. These disorders may occur during the stages of proliferation, migration, or organization of the cortex. The clinical manifestations of these disorders vary considerably, and they depend largely on the stage of arrest. Disorders of cortical formation are an important cause of developmental delay and epilepsy. MR imaging is a valuable tool in the diagnosis of these disorders.1–6
The purpose of this article was to review the embryologic stages of the cerebral cortex, illustrate the classification of the disorders of cortical formation, and finally describe the main MR imaging features of these disorders.
Embryology
The development of the cerebral cortex is divided into 3 stages: cell proliferation, cell migration, and finally cortical organization. During cellular proliferation, both the neuronal and glial precursors are generated, whereas during migration, these cells travel from the proliferative zone to their final designated destination. Formation of the cellular network takes place during the stage of cortical formation.1–3,7
Neural proliferation takes place between the second and fourth months of gestation. The neurons and glial cells develop from their neuroblast precursors in the ventricular and subventricular zones. Early, the neuronal-glial stem cells predominately divide to form additional stem cells. Later, stem cell division becomes asymmetric so that 1 daughter cell is postmitotic while the other remains as stem cell. Anomalies of neuronal proliferation may result in conditions characterized by too many, too few, or abnormal neurons.7,8
Neuronal migration occurs between the third and fifth months of gestation. Migration of the neurons occurs from the germinal zone to their final destination, close to the pial surface in 6 successive waves. As each wave of neuroblasts migrates, it passes to the pial extent of the cytoplasmic process before separating from it. Therefore, the first neurons to migrate occupy the deepest portion within the cerebral cortex, whereas those migrating later occupy the more superficial portions of the cortex. Migration is regulated by radial glial fibers and mediators. Disturbance of the guidance mechanism or the timely detachment of the migrating neurons in the cerebral cortex results in migration abnormalities. Disorders of neuronal migration are due to undermigration, overmigration, or ectopic migration of the neurons.3,8
Cortical organization, which appears to depend on normal neuronal migration, starts at 22 weeks of fetal gestation and continues until 2 years of age. Neurons differentiate into several cell types (pyramidal cells, stellate cells), which organize into horizontal laminar aggregates and vertical columns, leading to the formation of the normal cortical cytoarchitectonic pattern. Disorders of cortical organization result in abnormalities of gyral formation and cortical organization.3,7,8
Genetics
Disorders of cortical formation are commonly caused by mutation in a specific gene that acts in a dominant or X-linked fashion. Several new genes and new mutations of known genes for disorders of cortical formation have been mapped or cloned.4,7 Patients with severe germline mutations, such as deletions and truncations, typically have severe phenotypes and somatic mosaic mutations and often have a mild phenotype. The mutation is present in some cells in mosaicism and in all cells in the germline mutation. The type of mutation is often as important as identifying the gene that has been mutated in determining the clinical and imaging phenotypes of the affected patients. The recognition of such mechanisms is of great importance in genetic counseling.1,4,7–9
Classification
The classification scheme of disorders of cortical formations evolves every few years on the basis of neurogenetic insight into the various syndromes. Barkovich et al in 19967 proposed a classification scheme for disorders of cortical formation, based on the first step at which the developmental process was disturbed. This classification was updated by Barkovich et al in 2001.10 Last, Barkovich et al in 20059 described the disorders of cortical malformations by the causative gene rather than by the clinical phenotype wherever possible. The Table shows a simplified form of the classification of disorders of cortical formation.
Disorders of cortical formation according to stages
These 3 major stages of cortical development are not temporally separate: Proliferation continues after migration begins and migration continues as organization commences. The cell resulting from abnormal proliferation often does not migrate or organized properly.
Disorders of cortical formation resulting from abnormal cell proliferation might result from too many or too few neurons or abnormal cells. Microlissencephaly results from too few neurons, hemimegalencephaly results from too many cells, and focal cortical dysplasia (FCD) results from abnormal cells. Disorders of neuronal migration include malformations such as classic lissencephaly, which results from arrest of neuronal migration; cobblestone lissencephaly, which develops from overmigration of the neurons; and heterotopia, which occurs as a result of ectopic neuronal migration. Disorders of cortical organization result from prenatal ischemia or infection. Polymicrogyria (PMG) results from injury of the superficial part of the brain, whereas schizencephaly is due to insult of the whole thickness of the brain.
Malformations of the proliferative stage extend from the ventricles to the pia mater. Those that result from abnormal cellular migration are located in the white matter and/or the cortex, and those caused by abnormal organization are limited to the cortex.1,2,9,10
Microlissencephaly/Microcephaly with a Simplified Gyral Pattern
Microlissencephaly/microcephaly with a simplified gyral pattern is characterized by profound microcephaly and abnormal sulcation. The patient presents with profound congenital microcephaly (head circumference <3 SD below normal for that age) without in utero injury. This disorder results from either decreased cell production or increased apoptosis (programmed cell death) in the germinal zone of the cerebral cortex. It is classified into microlissencephaly (severe form) and microcephaly with a simplified gyral pattern (mild form).
Microlissencephaly is characterized by a smooth cortical surface with a thickened cortex (>3 mm) and is usually associated with other congenital anomalies (Fig 1). Microcephaly with a simplified gyral pattern is a mild form of the malformation with too few sulci and normal cortical thickness (3 mm) and is usually an isolated anomaly.1,11,12

Microlissencephaly. A, Sagittal T1-weighted image shows no sulcation in the cerebrum, a hypoplastic cerebellum, and a small brain stem with Dandy-Walker syndrome. B, Axial T1-weighted image shows agyria of the cortex.
Hemimegalencephaly
Hemimegalencephaly is characterized by enlarged and dysplastic hamartomatous overgrowth of part or all of the cerebral hemisphere. It results from too many neuromas or decreased apoptosis. It may be an isolated anomaly; a total hemimegalencephaly with enlargement of the ipsilateral cerebellum and brain stem; or associated with syndromes such as epidermal nevus syndrome, proteus syndrome, unilateral hypomelanosis of Ito, neurofibromatosis type I, Klippel-Trenaunay syndrome, and tuberous sclerosis.
Patients with hemimegalencephaly show moderate-to-marked enlargement of 1 or part of a cerebral hemisphere. The cerebral cortex may be normal or dysplastic (PMG, lissencephaly, or heterotopia). There is an indistinct cortical–white matter junction, with variable degrees of abnormal T1 and T2 prolongation of the white matter due to heterotopia and astrocytosis. The lateral ventricles are enlarged with a characteristic shape of the frontal horns that appears straight and pointed anteriorly and superiorly (Fig 2).13–17
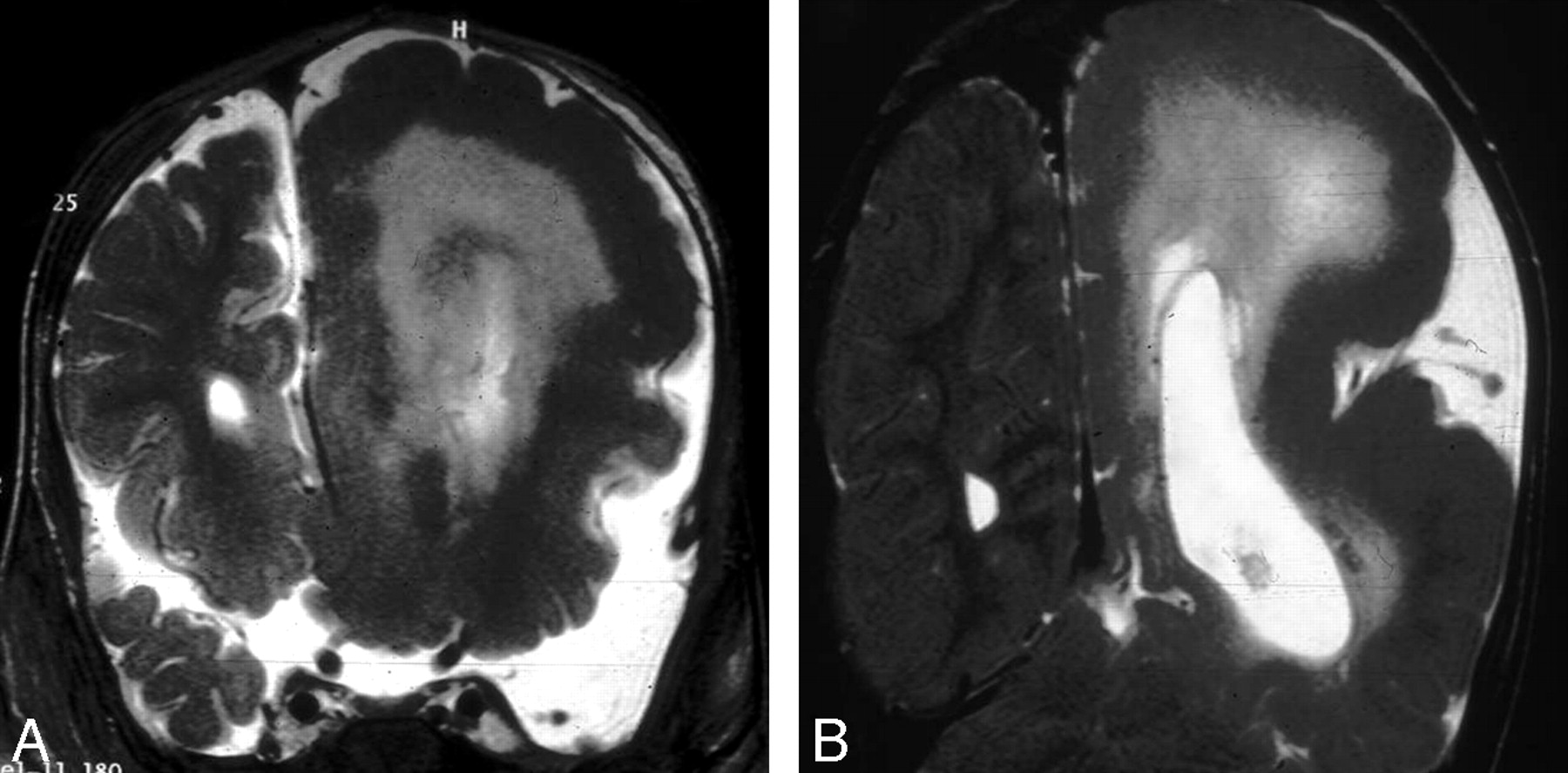
Hemimegalencephaly. A and B, Coronal T2-weighted images show an enlarged left cerebral hemisphere, a dilated left lateral ventricle, and a thickened cerebral cortex. Courtesy of Dr R. Zimmerman, Philadelphia.
FCD
FCD is a heterogeneous group of lesions characterized by the presence of abnormal neurons and glial cells within a localized region of the cerebral cortex. Patients usually present with intractable seizures. FCD encompasses a spectrum of malformations ranging from mild forms of cortical disruption without cellular abnormality to severe forms with marked cortical dyslamination, large bizarre cells, and astrocytosis of the cerebral cortex.
Taylor FCD is characterized by cytoarchitectural disarray of the cortex caused by large bizarre disoriented neurons together with the presence of balloon cells in the subcortical white matter and the cortex. The cortical neurons are increased in number and abnormally distributed. Balloon cells possess the characteristics of both neurons and astrocytes.
FCD appears as a localized area of cortical thickening with an indistinct gray-white matter junction. There is macrogyria and abnormally widened or deep sulci and a subcortical linear, curvilinear, radial, or funnel-shaped focus of abnormal signal intensity extending from the gray-white matter junction to the superolateral margin of the lateral ventricle. These foci show low signal intensity on T1-weighted images and high signal intensity on T2-weighted images (Fig 3). FCD should be differentiated from gliomas. A frontal location is in favor of FCD, whereas a temporal location is suggestive of neoplasm. High signal intensity on T2-weighted images is less distinct in FCD than in tumors because the main portion of cortical dysplasia lesions is located within the gray matter and is infrequently associated with edema or gliosis. Gliomas are associated with some degree of mass effect and might show a degree of enhancement after contrast injection.17–23

Focal cortical dysplasia. A and B, Axial and coronal T2-weighted images show focal cortical thickening in the right frontal lobe. Courtesy of Dr R. Zimmerman, Philadelphia.
Classic (Type I) Lissencephaly (4-Layer Lissencephaly)
Patients with classic lissencephaly may have a smooth brain surface in the complete form, or more commonly, they have a smooth surface with some gyral formation along the inferior frontal and temporal lobes in the incomplete form. This anomaly results from arrest of the migration process. Patients present with seizures and developmental delay in the complete form or complex seizures, hypotonia, microcephaly (50%), and facial dysmorphism (30%) in the incomplete form.
The malformation is characterized by complete agyria in the complete form or parieto-occipital agyria with frontotemporal pachygyria in the incomplete form. The cortex is thick because it encompasses radial columns of the arrested cells. The subcortical white matter is thin, with a lack of the normal gray-white matter interdigitation. There is a circumferential band of high signal intensity on T2-weighted images, most prominent in the parieto-occipital cortex, corresponding to a sparse cell zone with increased water content. Cerebral configuration is oval or hourglass with shallow Sylvian fissures due to lack of or incomplete operculization (Fig 4).
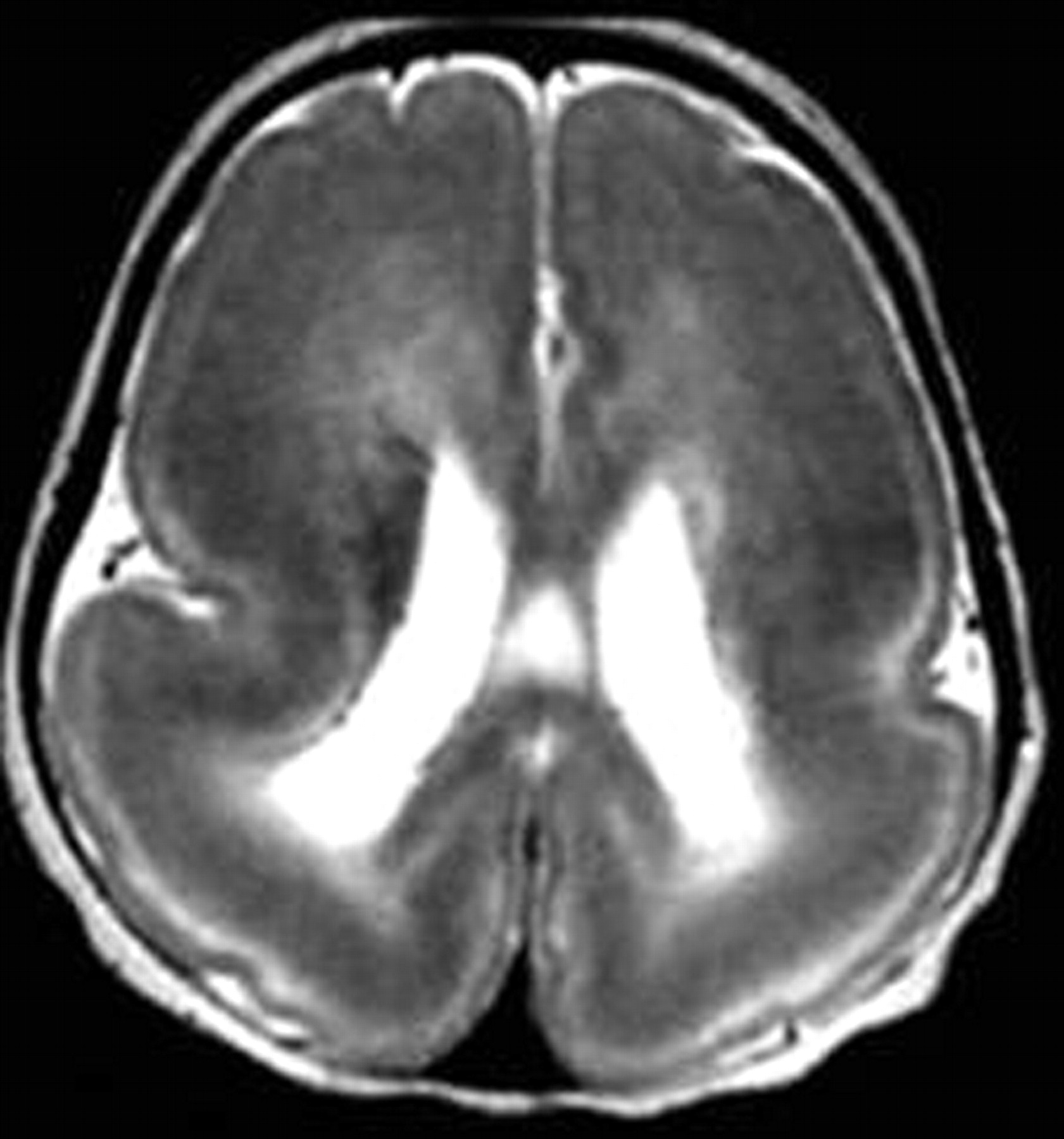
Complete lissencephaly. Axial T2-weighted image shows complete absence of sulci with a thick cortex, a shallow Sylvian fissure, and a circumferential band of high signal intensity in the parietooccipital cortex.
The malformation has several subtypes based on the underlying genetic abnormality. The LIS1 gene mutation is equal in males and females, with gyral abnormalities mostly in the form of parieto-occipital agyria and, less commonly, frontal pachygyria. The doublecorten gene is usually seen in females, with the gyral abnormalities mainly in the frontal lobes. The relin gene is a rare form of lissencephaly, with a small cerebellum, hypoplastic brain stem, and mild reduction of the cortical gyri. The ARX gene is a form of lissencephaly that is characterized by frontal pachygyria and parieto-occipital agyria, agenesis of the corpus callosum, and ambiguous genitalia.1,24–26
Cobblestone (Type II) Lissencephaly (Congenital Muscular Dystrophy)
Cobblestone lissencephaly is characterized by a nodular brain surface, ocular anomalies, and congenital muscular disorders. Cobblestone cortex results from overmigration of the neuroblasts and glial cells beyond the external glial limitations into the subarachnoid space (Fig 5). It includes a spectrum of anomalies with Walker-Warburg syndrome (WWS) being the most severe form; Fukuyama congenital muscular dystrophy (FCMD), the mildest form; and muscle-eye-brain (MEB) disease, the intermediate form.27–30

Cobblestone lissencephaly. A and B, Axial and coronal T2-weighted images show an irregular nodular cortex with hypomyelination of the white matter.
WWS is due to an abnormality of chromosome 9q34. It is characterized by profound hypotonia, severe ocular abnormalities, progressive macrocephaly, and posterior cephalocele. There is diffuse cobblestone lissencephaly, with completely unmyelinated white matter. It is associated with fused colliculi, a small pons, a dysmorphic mesencephalon, a dorsal pontomedullary kink, vermian hypogenesis, and cerebellar hypoplasia. Uni- or bilateral microphthalmia and retinal dysplasia (small and dysplastic globe) are commonly seen. Hydrocephalus and posterior cephalocele are common (Fig 6).27–31
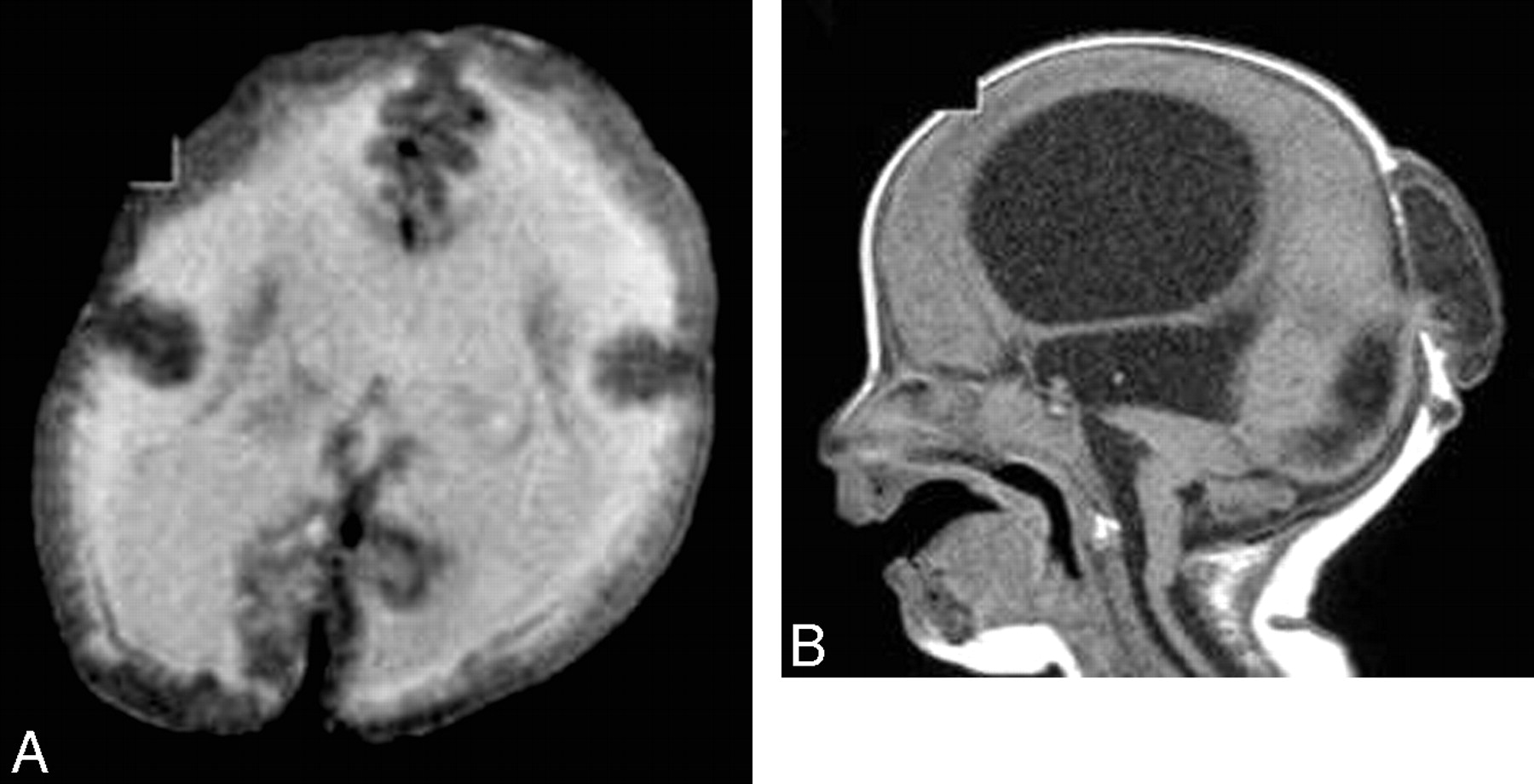
WWS. A, Axial T2-weighted image shows a cobblestone cerebral cortex with hypomyelination of the white matter. B, Sagittal T1-weighted image shows hydrocephalus, an occipital encephalocele, a pontomedullary kink, and a small pons.
MEB disease is due to an abnormality of chromosome 1p32–p34 and is commonly seen in Finland. This disorder presents with mild hypotonia, mild ocular anomalies, and developmental delay. There is little undersulcation of the frontal lobes with hypomyelination of the white matter, fused colliculi, hypoplasia of the pons with a longitudinal anterior cleft, vermian hypoplasia, cerebellar PMG, and cysts. The globe is small with subretinal fluid and retinal dysplasia. Absent septum pellucidum, callosal hypogenesis, and/or hydrocephalus might be seen (Fig 7).27–31

MEB syndrome. A, Axial T2-weighted image shows undersulcation of the frontal lobe, hypomyelination of the frontal white matter, and dilated lateral ventricles. B, Axial T2-weighted image shows multiple small cerebellar cysts, with a small pons and bilateral temporal arachnoid cysts.
FCMD is due to an abnormality of chromosome 9q31–33 and is primarily seen in the Japanese. It is the most characteristic congenital muscular dystrophy, with an intermediate clinical picture between WWS and FCMD. There is PMG in the frontal region and cobblestone lissencephaly in the parietooccipital region. Delayed myelination, if present, proceeds from the peripheral subcortical region centrally, the reverse of normal myelination. There are colliculi fusion cerebellar PMG and cysts. Callosal anomalies and cephalocele are unusual. Some patients have diffuse cortical and ocular dysplasia .32,33
Heterotopia
Heterotopia represents collections of normal neurons located in abnormal locations, anywhere from the subependymal region to the cerebral cortex. They result from the arrest of neuroblast migration. Patients commonly present with epilepsy and possible delayed milestones and/or mental retardation. Heterotopia may be associated with pachygyria, agenesis of the corpus callosum, Chiari II malformation, arachnoid cyst, schizencephaly, and cephalocele.1,2,6,34,35
Periventricular (Subependymal) Heterotopias
Periventricular (subependymal) heterotopias (PVH) are located in close proximity to the ventricular wall. They are commonly seen in the region of the trigone and occipital horns of the lateral ventricles. PVH are usually bilateral with predilection for the right cerebral hemisphere due to later migration of the right-sided neuroblasts. They appear as round or oval nodules, are isointense to the normal gray matter on all pulse sequences, and do not enhance after contrast injection. They may lie in the wall of the ventricle and project into the ventricular lumen or lie within the periventricular white matter. Mild ventricular dilation might be seen. PVH can be subdivided into unilateral focal, bilateral focal, and bilateral diffuse groups (Fig 8).1,2,34–36

PVH. A and B, Coronal T1-weighted images show a few small periventricular nodules, isointense to the gray matter, along the lateral ventricular wall.
Subcortical Heterotopias
Subcortical heterotopias (SCH) are located within the subcortical or deep white matter and are always seen contiguous to the overlying cortex or the underlying ventricular system. The affected hemisphere may decrease in size as a result of the decreased volume of the neurons. The overlying cortex appears thin with shallow sulci. SCH are either nodular, curvilinear, or mixed form.
Nodular SCH appear as nodules that extend from the ventricular surface outward into the white matter without continuity with the cerebral cortex. They are variable in size from small lesions of less than a centimeter to large lesions of several centimeters that may occupy almost 1 cerebral hemisphere. Nodular SCH appear as a large mass lesion on 1 side of the brain.
Curvilinear SCH consist of swirling heterogeneous curvilinear masses of gray matter that have an appearance that closely resembles convolutions of the cortex, extending from the cortical surface into the white matter. They may contain blood vessels and CSF. The blood vessels course within the layer of the gray matter, and the CSF is seen between the gray matter convolutions. When both occur, the vessels seem to course within the fluid. The blood vessels and fluid seen within heterotopia are due to abnormal communication with the subarachnoid space (Fig 9).

Curvilinear SCH. Axial T2-weighted MR image shows curvilinear heterotopias in the right cerebral hemisphere that are associated with its decrease in size.
In mixed SCH, nodules are seen in the deep part of the brain adjacent to the lateral ventricle, and the curvilinear convolutions are seen in the superficial part (Fig 10).34–37

Mixed SCH. Axial T2-weighted MR image shows a large nodule adjacent to the lateral ventricle with a signal intensity similar to that of the gray matter and curvilinear convolutions in the superficial part.
MR spectroscopy can differentiate heterotopia from low-grade glioma. The metabolites appear to be similar to those of normal brain in heterotopia, whereas loss of N-acetylaspartate and increase of choline were observed in low-grade gliomas.38–39
Band (Laminar) Heterotopia
Band heterotopia is a rare anomaly seen predominantly in females and may be familial, with X-linked dominant inheritance. It results from an early arrest of neuronal migration and consists of a smooth layer of the gray matter that often follows the curvature of the overlying cortex. On MR imaging, it shows the characteristic 3-layer cake (continuous double cortex) with the cortex and bilateral symmetric circumferential subcortical layer of band heterotopia separated from each other by a thin white matter band. The cortex may be relatively normal or pachygyric.2–4,40
PMG
PMG is characterized by many small prominent convolutions separated by shallow sulci with an irregular appearance of the cortical surface and cortical white matter. It results from prenatal infection, ischemia, or exposure to toxins or chromosomal abnormality. The malformation is characterized by a heterogeneous collection of neurons and derangement of the normal 6-layer lamination of the cortex. The sulci may be obliterated and bridged by fusion of their superficial cellular layers, particularly the molecular one. The anomaly can be unilateral or bilateral, small or large, symmetric or asymmetric. It is commonly located in regions adjacent to the Sylvian fissures (ie, the posterior frontal lobe, the superior temporal lobe, and the inferior parietal lobe).2–4,41
PMG is characterized by small fine undulating gyri, very similar to an undulating cortical ribbon, which cannot be detected on imaging. The cortex is also slightly thickened (5–7 mm) with an abnormal increased T2 signal intensity of the underlying white matter, which is noted in 25% of cases.2,41 When PMG is due to congenital infection, the areas of T2 prolongation are present bilaterally in the cerebral white matter. An anomalous vein is commonly seen in the region of PMG.
There are 2 distinct patterns of PMG: Pattern I is characterized by a small fine undulating cortex with normal cortical thickness (3–4 mm). This pattern is seen in the anterior frontal region, mainly in infants younger than 12 months. There is an associated layer of T2 prolongation of the underlying white matter. Pattern II has a bumpy cortical appearance with less undulation. The cortex is abnormally thickened (5–7 mm) with an irregular cortical–white matter junction. This pattern is noted in infants older than 18 months. It involves mainly the frontal, parietal, and perisylvian regions. Differentiation between the cortex and underlying white matter is poor on T1-weighted images in this latter pattern (Fig 11).42

PMG. Axial T2-weighted image shows microgyria with normal cortical thickness that is associated with the high signal intensity of the white matter.
PMG Syndromes
These syndromes affect the cognitive regions (frontal, frontoparietal, perisylvian, and parieto-occipital regions), and patients usually present with epilepsy and cognitive delay. Additional symptoms depend on the specific region affected.43
Bilateral perisylvian PMG (congenital bilateral perisylvian syndrome) consists of primitive Sylvian fissures, primitive draining veins, and dysplastic operculum. It may be sporadic or familial. It usually presents with pseudobulbar palsy, epilepsy, cognitive impairment, and congenital arthrogryposes. On MR imaging, the Sylvian fissures are wide, deep, and underdeveloped and extend farther back. An anomalous vein is seen in the unfolding dysplastic cortex. The opercula are dysplastic due to symmetric involvement with PMG and pachygyria. It may be seen in families linked to the locus on Xq28.43–45
Bilateral frontal PMG presents with cognitive and motor delay and spastic quadriparesis with epilepsy. There is symmetric PMG extending from the frontal poles posteriorly to the precentral gyrus and inferiorly to the frontal operculum. All cases are sporadic and patients may have consanguineous parents.46
Bilateral frontoparietal PMG is commonly seen in the Middle East and India. Patients present with developmental delay, mild spastic quadriparesis, impaired language, and epilepsy with cerebellar dysfunction. There is characteristic bilateral symmetric PMG in the frontoparietal regions, with decreased severity anteriorly to posteriorly. The white matter is thin, with areas of T2 prolongation. The ventricles are enlarged, and the pons and cerebellum are small. It is autosomal recessive, with the locus identified on 16q12.2.47
Bilateral parasagittal parieto-occipital PMG presents with seizures and mental retardation; however, neurologic deficits are often absent. Bilateral PMG is seen in the parasagittal and mesial aspects of the parietooccipital cortex. All cases are sporadic.48
Bilateral generalized PMC presents with cognitive and motor delay of variable severity with seizures. There is symmetric generalized PMG, often most prominent in perisylvian regions. Most cases are presumed to be autosomal recessive.49
Schizencephaly
Schizencephaly is a cleft lined with gray matter and connecting the subarachnoid CSF spaces with the ventricular system. Schizencephaly results from injury involving the entire thickness of the developing hemisphere during cortical organization. The injuries are due to prenatal infection, ischemia, or chromosomal abnormalities. Clinical manifestations of schizencephaly most often include varying degrees of developmental delay, motor impairment, and seizures. It might be associated with septo-optic dysplasia, optic nerve hypoplasia or the absence of septum pellucidum, pachygyria, PMG, heterotopia, and arachnoid cysts.
Schizencephaly appears as a cleft filled with CSF and extending medially from the subarachnoid space into the lateral ventricle. The wall of the cleft is lined with dysmorphic gray matter. The gray matter sometimes extends into the lateral ventricle in the form of subependymal heterotopias. The cleft may be small or large, unilateral or bilateral. The anomaly may be of the open-lip or closed-lip type. Closed-lip schizencephaly is characterized by gray matter–lined lips, which are in contact with each other (type 1). Open-lip schizencephaly has separated lips and a cleft of CSF extending to the underlying ventricles (type 2) (Fig 12).50–52

Schizencephaly. A, Coronal T2-weighted image shows a closed-lip right-sided schizencephalic defect lined by pachygyria. (Courtesy of Dr R. Zimmerman, Philadelphia.) B, Coronal T2-weighted image shows a wide CSF-filled cleft connecting the left lateral ventricle with the subarachnoid space, which is lined with gray matter parenchyma.
Conclusion
MR imaging is an important tool for the diagnosis of disorders of cortical formation. It is essential to demonstrate the morphology, distribution, and extent of these disorders. Moreover, it can identify the associated congenital anomalies and related syndromes and suggest the genetic abnormalities.
Acknowledgments
We thank Robert Zimmerman, MD, Professor of Radiology at Children's Hospital of Philadelphia, for his help with some figures of this article.
Footnotes
Paper previously presented at: Annual Meeting of the Radiological Society of North America, November 26–December 1, 2006; Chicago, Ill.
References
- Received April 24, 2008.
- Accepted after revision May 26, 2008.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Embryology
- Classification
- Microlissencephaly/Microcephaly with a Simplified Gyral Pattern
- Hemimegalencephaly
- FCD
- Classic (Type I) Lissencephaly (4-Layer Lissencephaly)
- Cobblestone (Type II) Lissencephaly (Congenital Muscular Dystrophy)
- Heterotopia
- Periventricular (Subependymal) Heterotopias
- Subcortical Heterotopias
- Band (Laminar) Heterotopia
- PMG
- Schizencephaly
- Conclusion
- Acknowledgments
- Footnotes
- References
- Figures & Data
- References
- Info & Metrics