
Abstract
BACKGROUND AND PURPOSE: In recent years, the discovery that mutations in the glial fibrillary acidic protein gene (GFAP) were responsible for Alexander disease (AD) brought recognition of adult cases. The purpose of this study was to demonstrate that MR imaging allows identification of cases of AD with adult onset (AOAD), which are remarkably different from infantile cases.
MATERIALS AND METHODS: In this retrospective study, brain and spinal cord MR imaging studies of 11 patients with AOAD (7 men, 4 women; age range, 26–64 years; mean age, 43.6 years), all but 1 genetically confirmed, were reviewed. Diffusion and spectroscopic investigations were available in 6 patients each.
RESULTS: Atrophy and changes in signal intensity in the medulla oblongata and upper cervical spinal cord were present in 11 of 11 cases and were the diagnostic features of AOAD. Minimal to moderate supratentorial periventricular abnormalities were seen in 8 patients but were absent in the 3 oldest patients. In these patients, postcontrast enhancement was also absent. Mean diffusivity was not altered except in abnormal white matter (WM). Increase in myo-inositol (mIns) was also restricted to abnormal periventricular WM.
CONCLUSIONS: Awareness of the MR pattern described allows an effective selection of the patients who need genetic investigations for the GFAP gene. This MR pattern even led to identification of asymptomatic cases and should be regarded as highly characteristic of AOAD.
Up to a few years ago, Alexander disease (AD) was known as a rare, genetically determined leukoencephalopathy affecting infants and children, characterized by macrocephaly, seizures, spasticity, and retarded psychomotor development, and leading to death in 2 months to 7 years.1 The diagnostic MR imaging features established by van der Knaap et al2 consisted of extensive white matter (WM) increased signal intensity on T2-weighted images, mainly in the frontal regions; a rim of periventricular T2 hypointensity; involvement of the basal ganglia, thalami, and brain stem; and postcontrast enhancement in the periventricular regions and scattered areas of the brain stem. These features allowed the diagnosis without biopsy, which was performed in the search for Rosenthal fibers, the histologic hallmark of the disease.2,3
Milder forms with spastic paraparesis, ataxia, or lower brain stem signs and juvenile (2–12 years of age) or adult onset (≥13 years) had been diagnosed as AD at postmortem examination because of the presence of Rosenthal fibers.4 After 2001, the discovery that mutations in the glial fibrillary acidic protein gene (GFAP) were responsible for the disease5,6 allowed recognition during life of juvenile or adult forms1,7–20 that have an MR imaging pattern remarkably different from that observed in children, with predominant focal involvement of the lower brain stem.
In the last 4 years, on the basis of the MR imaging findings, we suggested the diagnosis of adult-onset AD (AOAD) in 11 patients, all but one genetically confirmed, and were struck by the constant involvement of the medulla oblongata and upper spinal cord. Therefore, we have analyzed retrospectively the imaging studies to verify the reliability of MR in making the diagnosis. The results of the genetic studies are also briefly reported.
Materials and Methods
Patients
All patients included in this study except 2 were admitted to our institute, either directly or referred from other centers for an undiagnosed neurologic disorder. One patient (patient 1) had a 7-year follow-up in another institution and was seen for a neuroradiologic consultation in our department. The first 2 patients had already been reported.17,18
In addition to the 10 patients in whom AOAD was genetically confirmed, an eleventh patient whose AOAD was not genetically confirmed who presented clinical and radiologic findings identical to those observed in the other 10 patients with AOAD was included in the present series because he also had autosomal dominant disease transmission. Lack of this support led to exclusion of another patient born to consanguineous parents.
The patients (7 men, 4 women; age range, 26–64 years) presented with history of brain stem disturbances, ataxia, spastic paraparesis, or bladder dysfunction, progressing for a period of a few months to 13 years (Table 1). Five patients were moderately to severely impaired in their daily activities (patients 2, 3, 4, 7, and 11), and 4 were still able to conduct a nearly normal life (patients 1, 6, 8, and 10). Two patients had a normal neurologic examination: patient 5 had an MR study as a work-up for a persistent unclassified headache, and patient 9 had an MR image in the follow-up studies for a subarachnoid hemorrhage of unknown origin. The unexpected MR imaging findings prompted genetic testing for AD.
Clinical features in 11 patients with AOAD
Imaging
All of the patients were submitted to an MR investigation as part of their clinical work-up. Therefore, this study was retrospective with some inhomogeneity of sequences. All of the patients gave written informed consent to imaging studies, which complied with institutional guidelines and regulations.
All MR studies (except those performed in patient 1) were performed on a 1.5T system (Avanto; Siemens, Erlangen, Germany). Brain MR imaging examinations were performed with axial proton-attenuation and T2-weighted sequence (TR, 3200 ms; TE, 11 ms and 126 ms; FOV, 230 × 200 mm; matrix, 384 × 270; section thickness, 5 mm with 1-mm gap), sagittal T1-weighted sequence (TR, 512 ms; TE, 9 ms; FOV, 230 × 230 mm; matrix, 256 × 218; section thickness, 5 mm with 1-mm gap), and coronal fluid-attenuated inversion recovery (FLAIR) sequence (TR, 8700 ms; TE, 121 ms; TI, 2500 ms; FOV, 240 × 192 mm; matrix, 320 × 180; section thickness, 5 mm with 1-mm gap). In addition, in a few subjects, sagittal 3-mm thick T1-weighted images, sagittal or axial FLAIR sequences, and sagittal or axial 3-mm thick T2-weighted sequences were obtained. Postcontrast (gadopentetate dimeglumine, Magnevist; Shering, Berlin, Germany) axial, sagittal, and coronal 5-mm thick T1-weighted sequences were obtained in 9 patients.
Cervical spine MR imaging studies were performed with sagittal T1-weighted sequence (TR, 600 ms; TE, 12 ms; FOV, 250 × 250 mm; matrix, 384 × 346; section thickness, 3 mm with 0.3-mm gap), sagittal T2-weighted sequence (TR, 3000 ms; TE, 104 ms; FOV, 240 × 240 mm; matrix, 384 × 288; section thickness, 3 mm with 0.3-mm gap), and axial T2*-weighted sequence (TR, 951 ms; TE, 22 ms; FOV, 210 × 158 mm; matrix, 256 × 173; section thickness, 4 mm with 1.2-mm gap). Postcontrast sagittal T1-weighted sequences were also obtained. Complete spinal cord examination with analogous parameters was obtained in 3 patients.
MR examinations obtained elsewhere less than 1 month before referral to our institute were not considered. Three patients (patients 1, 3, and 10) had multiple examinations with a maximum interval of 7 years.
Six patients (patients 3, 4, 5, 6, 7, and 11) underwent MR imaging with an axial single-shot, spin-echo, echo-planar diffusion-weighted sequence (TR, 3000 ms; TE, 87 ms; FOV, 230 × 230 mm; matrix, 128 × 128; b-values, 0–500–1000 seconds mm−2; 3 orthogonal directions; thickness, 5 mm with 1.5-mm gap), from which mean diffusivity maps were generated. For patients 3 and 6 (who had extensive supratentorial WM abnormalities), 8 circular regions of interest (ROIs) (area, 2.2 cm2) for measurement of diffusivity were positioned in normal-appearing and abnormal-appearing frontal and parietal WM. For patients 4, 5, 7, and 11, 4 ROIs were positioned in deep frontal WM and deep parietal WM.
Six patients (patients 3, 6, 7, 8, 10, and 11) were studied with proton MR spectroscopy by means of a short echo-time multivoxel point-resolved spectroscopy sequence (TR, 1500 ms; TE, 30 ms; FOV, 160 × 160 mm; matrix, 16 × 16; thickness, 15 mm). The section was positioned on the centra semiovalia. We determined the relative amplitudes of the peaks of choline, N-acetylaspartate (NAA), and myo-inositol (mIns) by using frequency-domain fitting and taking the creatine peak as a reference value.
In 3 patients (patients 3, 8, and 10), we attempted to acquire short echo-time spectra from a single 16 × 18 × 33-mm voxel, positioned on the medulla oblongata and including the lower third of the pons; 2 of these attempts failed because of high-susceptibility artifacts and motion caused by CSF pulsatility.
Reference values for diffusion and spectroscopy were obtained from repository data of healthy subjects studied with the same sequences on the same scanner (n = 6 and 50.3 ± 7.3 years for diffusion, and n = 11 and 43.3 ± 9.0 years for spectroscopy). The spectrum obtained in the medulla oblongata was compared with spectra acquired in the pons in the control group. Diffusion and spectroscopy measurements from the patients (except patients 3 and 6) were compared with control values by means of 2-tailed unequal-variance t tests. Patients 3 and 6, who had extensive supratentorial WM abnormalities, were considered separately, and values falling within 2 SDs of the control group were interpreted as unaltered. In all comparisons, values from the left and right hemispheres were averaged and considered together.
All of the examinations were reviewed by 2 neuroradiologists (L.F., M.S.) blind to the clinical severity of the patients’ conditions, not to the genetic diagnosis, which, in nearly all cases, had been made on their indication after the first reading of the MR images.
The MR images were reviewed for atrophy and changes in signal intensity in the supratentorial regions, brain stem, cerebellum, and spinal cord. Involvement of the WM of the cerebral hemispheres was graded on the basis of thickness of the periventricular band of signal intensity changes as a thin or large band. No volumetric studies were available, but a conjoint evaluation was made to graduate medullary atrophy from 0 (no atrophy) to 2 (1 = mild to moderate, 2 = severe atrophy). Medullary abnormalities of signal intensity were also jointly evaluated in consideration of their extension and conspicuousness in the different sequences; they were also visually graded 0 = absent, 1 = mild to moderate, and 2 = severe. However, in consideration of the difficulties in grading atrophy and signal intensity changes in the absence of measurements, the simple presence or absence of these parameters was also registered. Contrast enhancement was evaluated for distribution and number of areas involved.
Genetic tests
Exons 1 to 9 of the GFAP gene, including flanking intronic sequences, were investigated by direct sequencing, the use of primers, amplification conditions, and a protocol already reported.21 The presence of each nucleotide variant thus detected in the present patients was tested in 50 unrelated healthy individuals (100 control chromosomes) to distinguish between disease-related mutations and polymorphisms.
Results
MR Imaging
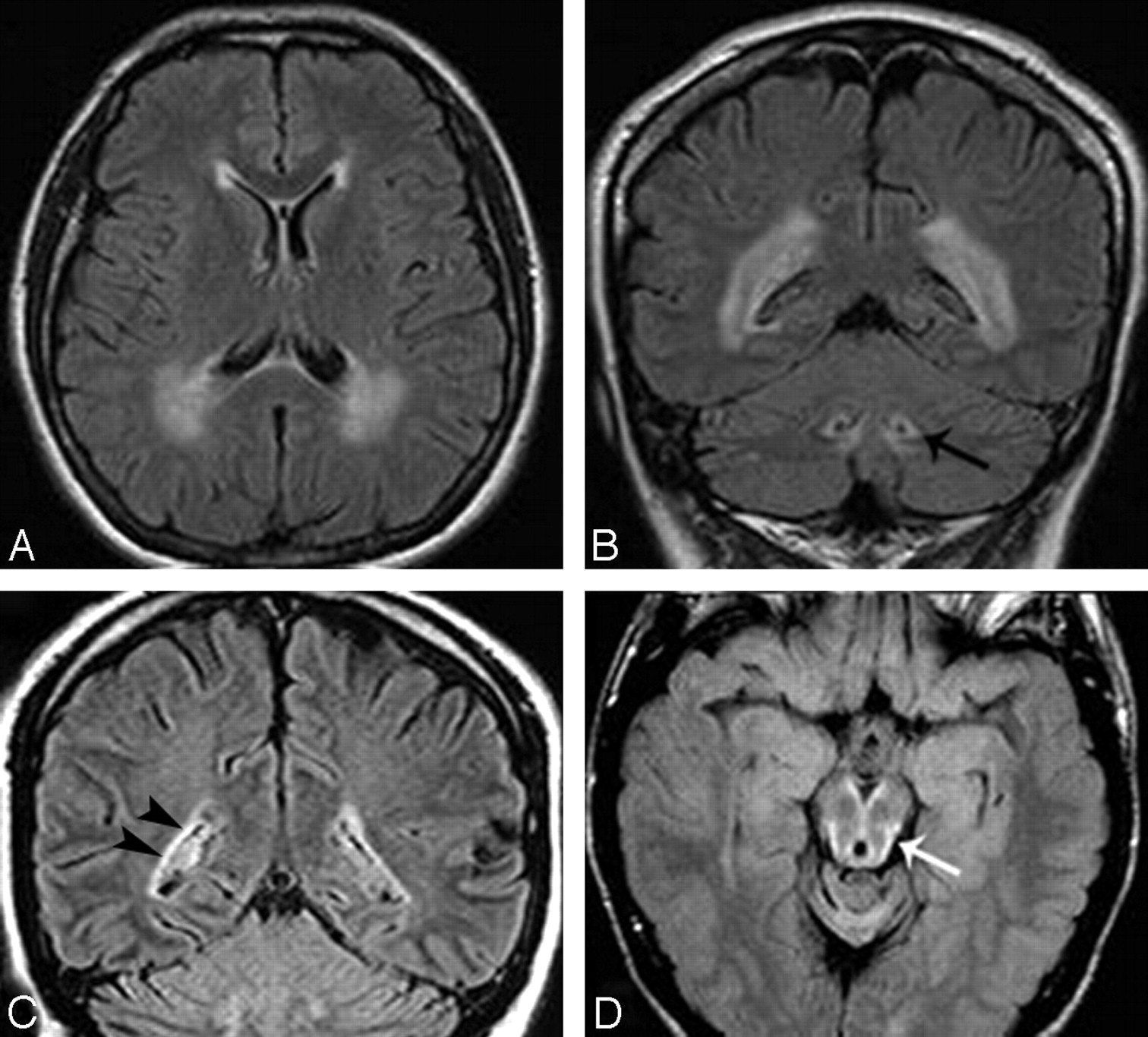
All patients had atrophy and signal intensity abnormalities of the medulla oblongata, extending caudally to the C1-C2 segments of the spinal cord (Table 2, Fig 1). Atrophy was mild to moderate in 4 patients and severe in 7. The changes in signal intensity sometimes selectively involved the medial lemniscus and the corticospinal tracts. The inferior cerebellar peduncles were extensively affected in 1 patient (patient 1) and only minutely involved in 4 (patients 3, 4, 8, and 11). The abnormalities did not extend cranially in the pons more than a few millimeters except in patient 11, who showed asymmetric extension to the middle third of the basis pontis. The middle and superior cerebellar peduncles were involved in 4 patients (patient 5 and patients 4, 6, and 11, respectively). In 5 patients (patients 3, 4, 6, 9, and 11), the midbrain was involved with a very subtle superficial rim of hyperintensity on proton attenuation and FLAIR images (Fig 2).

A, T2-weighted sagittal section in patient 9 shows thinning of the medulla oblongata and spinal cord with hyperintensity in the medulla; arrow indicates its posterior concave profile. B–D, Axial T2-weighted sections (thickness 3 mm) in the same patient demonstrate increased signal intensity involving the hilum of the dentate nuclei (arrow on the left side), the medial lemniscus (arrowhead, B), and the corticospinal tracts (arrowheads on the left side) in the medulla (C and D). In patient 11 (E), axial spinal cord T2-weighted section at the C1-C2 level shows severe atrophy with mild changes in signal intensity.

Axial and coronal FLAIR images in patient 6 show signal intensity changes prevalent in the cerebral posterior periventricular regions (A and B) and involvement of the hilum of the dentate nuclei (arrow on the left side, B). In patient 11, coronal FLAIR image (C) shows a thin bilateral band of periventricular hyperintensity (arrowheads on the right) not recognizable on T2-weighted images (not shown). In patient 3, midbrain peripheral rim of hyperintensity (arrow) is seen only on FLAIR section (D).
MR imaging findings in 11 patients with AOAD
In the upper part of the spinal cord, the signal intensity abnormalities appeared to be located centrally and in the lateral columns. In 4 patients, mild changes in signal intensity extended below C2 and faded away between C3 and C6. In all patients, the middle and lower cervical spinal cord segments were also atrophic. In patients 7 and 11, the atrophy was more severe than in the medulla. In the 3 patients who underwent complete spinal cord examination, mild atrophy without changes in signal intensity was present below the cervical segment.
In 8 patients, abnormalities in signal intensity were present in the hilum of the dentate nuclei; in patient 3, they were associated with abnormalities in the cerebellar WM. In patient 10, slight hyperintensity was present in the entire cerebellum on proton attenuation and FLAIR images. In patients 3 and 10, mild to moderate infratentorial and supratentorial atrophy was present.
In all but 3 patients, the cerebral periventricular WM showed some abnormalities in signal intensity (Fig 2). They were more extensive in 2 patients, also recognizable as hypointensity on T1-weighted images. Preponderant frontal involvement was seen in 3 patients (patients 2, 3, and 5). However, in 3 other patients (patients 4, 6, and 9), involvement of the WM was preponderant in the posterior regions. The irregular ventricular profile, described by van der Knaap15 as the “garlands,” was present in patient 1 and questionable in patient 3.
Four patients had a cyst, usually small, of the septum pellucidum. No patient had abnormalities in the basal ganglia.
Patchy areas of postcontrast enhancement, mainly in the brain stem and cerebellum, were demonstrated in 5 of 10 patients (Fig 3). In patient 3, patchy areas of enhancement were also seen around the frontal horns.

In patient 3, sagittal T1-weighted section (A) shows severe atrophy of the medulla oblongata (arrow) and spinal cord. Moderate cerebellar and cerebral atrophy is also present. Postcontrast axial sections (B and C) demonstrate 2 small areas of abnormal enhancement (arrows).
The previous MR examinations of patients 1, 3, and 10 were also reviewed. In patient 1, a series of MR studies demonstrated development of progressive atrophy of the medulla oblongata with signal intensity changes and abnormal enhancement present since the first examination. The cervical spinal cord was already thin at the first observation.17 In patient 3, brain stem atrophy and WM abnormalities progressed for 7 years. Postcontrast enhancement persisted for the last 3 years. In patient 10, no significant differences were seen on 3 examinations obtained in 1 year.
Diffusion-Weighted Imaging
In patient 3, who had extensive supratentorial WM abnormalities, diffusivity was unaltered in normal-appearing frontal and parietal WM. In abnormal frontal WM (data presented as mean ± SD), diffusivity was increased by 98% (1.48 × 10−3 mm2 s−1 versus [0.75 ± 0.04] × 10−3 mm2 s−1). In abnormal parietal WM, diffusivity was increased by 27% (1.02 × 10−3 mm2 s−1 versus [0.80 ± 0.05] × 10−3 mm2 s−1).
In patient 6, who had WM abnormalities prevalent in the posterior regions, diffusivity was unaltered in normal-appearing frontal and parietal WM. In abnormal frontal WM, diffusivity was increased by 26% (0.94 × 10−3 mm2 s−1 versus [0.75 ± 0.04] ×10−3 mm2 s−1). In abnormal parietal WM, diffusivity was increased by 34% (1.07 × 10−3 mm2 s−1 versus [0.80 ± 0.05] × 10−3 mm2 s−1).
In the remaining 4 patients, who had mild or no WM abnormalities, diffusivity was not significantly increased in the frontal ROIs ([0.78 ± 0.06] × 10−3 mm2 s−1 versus [0.75 ± 0.04] × 10−3 mm2 s−1; P = .5) and in the parietal ROIs ([0.87 ± 0.05] × 10−3 mm2 s−1 versus [0.80 ± 0.05] × 10−3 mm2 s−1; P = .07).
MR Spectroscopy
In patient 3, in normal-appearing frontal WM, the relative amplitude of the mIns peak was increased by 67% (0.53 versus 0.32 ± 0.07), and those of the choline and NAA peaks were unaltered (data presented as mean ± SD). In abnormal frontal WM, the relative amplitude of the mIns peak was increased by 132% (0.74 versus 0.32 ± 0.07), and those of the choline and NAA peaks were unaltered. In normal-appearing parietal WM, the relative amplitudes of all peaks were unaltered. In abnormal parietal WM, the relative amplitude of the mIns peak was increased by 130% (0.86 versus 0.37 ± 0.10), and those of the choline and NAA peaks were unaltered. In the voxel positioned on the medulla oblongata, the relative amplitudes of the 3 peaks were unaltered.
In patient 6, in all considered regions, with either normal or abnormal-appearing WM, the relative amplitudes of the 3 peaks were unaltered. With regard to mIns particularly, no differences were found in the frontal (mIns relative amplitude of 0.25 in normal-appearing WM and 0.32 in abnormal WM versus 0.32 ± 0.07) and parietal regions (mIns relative amplitude of 0.24 in normal-appearing WM and 0.54 in abnormal WM versus 0.37 ± 0.10).
With respect to controls, no significant difference was found for any metabolite in the remaining 4 patients in the frontal (mIns relative amplitude 0.32 ± 0.02 versus 0.32 ± 0.07; P = .8) and parietal WM (mIns relative amplitude 0.37 ± 0.09 versus 0.37 ± 0.10; P = .9).
Genetic Studies
All of the patients but 1 carried heterozygous missense nucleotide changes of the GFAP gene, which were absent in 200 control chromosomes (Table 3). In particular, 3 patients (patients 3, 6, and 7) harbored mutations already reported in previous cases, whereas 7 patients had novel GFAP mutations. Patient 11 did not show any apparently causative mutation. He harbored a p.D157N missense variant, a rare polymorphism already reported in 1 patient with AD and his unaffected father,1 and control subjects1,22 though not found in our control chromosome set.
Missense mutations of the GFAP gene in 11 patients with AOAD
Discussion
The diagnosis of AOAD is usually proposed on the basis of MR imaging findings. Although the association of ataxia with palatal tremor may suggest AOAD,19 the clinical presentation is usually not sufficiently characteristic. When, in addition to atrophy and signal intensity changes in the medulla oblongata, periventricular WM abnormalities are present, the diagnosis of a leukodystrophic process should be entertained. However, when the MR findings are limited to the brain stem-spinal cord junction, a diagnosis requires that the neuroradiologist be aware of the characteristic features of AOAD. Although in recent years several single case reports and small series of mixed juvenile and adult patients with AD have shown the predominant lower brain stem involvement,1,7–20 no detailed analysis of a large number of AOAD has been reported. It is peculiar that in the period of collection of our AOAD series, no juvenile cases came to our observation. The MR imaging pattern of juvenile cases14,15 is very similar to that observed in adults and is remarkably different from that of the classic infantile form.2
In the radiologic differential diagnosis, several disorders occasionally need to be considered.17 Before development of atrophy, the diagnosis of brain stem glioma may be entertained. Multiple sclerosis, actually the diagnosis that a few patients referred to our institute were carrying, could be considered. Limitations of abnormalities to the medulla oblongata made this diagnosis unlikely even in patients who also had periventricular T2 hyperintensities with a pattern, however, different from that seen in multiple sclerosis. Viral encephalomyelitis from enterovirus 71 may present MR abnormalities involving the brain stem, dentate nuclei, and spinal cord,23 but a clinical history of acute infection makes these patients completely different from those with AOAD.
Persistent enhancement may suggest a granulomatous or histiocytic disorder. Erdheim-Chester disease, which is a non-Langerhans cell histiocytosis that may affect the brain stem and cerebellum, usually involves the pons, not the medulla oblongata.24 Behçet disease, instead, has the midbrain as the preferential target in the brain stem.25 Various mitochondrial disorders, particularly Leigh syndrome, often affect the medulla but also the pontine tegmentum, midbrain, subthalamic nuclei, and putamina.26 Leukoencephalopathy with involvement of the brain stem and spinal cord and elevated lactate may occasionally be considered.17,27 However, involvement of the brain stem is more extensive than that seen in AOAD, and the characteristic changes along the intrapontine fibers of the trigeminal nerve make the differential diagnosis easy.
Because of age of onset and slow evolution of AOAD, degenerative disorders that may involve the brain stem and cerebellum could also be considered, but progressive supranuclear palsy, multisystem atrophy with cerebellar predominance, and various spinocerebellar ataxias have different MR features and are easily excluded.
In our experience, the MR imaging pattern observed in our series should first suggest the diagnosis of AOAD even in the absence of clinical information and in asymptomatic subjects. Indeed, we found pathogenic GFAP mutations in 10 patients. Patient 11, with no pathogenic mutations, had only a rare polymorphism. He was included because of suggestive clinical features (including palatal myoclonus), typical MR pattern, and a father who died of a similar neurologic disorder with identical MR findings. A patient with AOAD, without genetic confirmation but with a positive family history, has already been reported,1 and a total of 5 cases are cited in the AD mutation data base (http://www.waisman.wisc.edu/alexander/).
Another sporadic patient we observed had ataxia, spasticity, and “typical” MR findings but was not found to have any GFAP mutation nor a compatible family history and therefore was not included in the present series. It is possible that he indeed has AOAD with promoter or intronic GFAP mutations or mutations in some other as yet unidentified gene associated with the disease. Alternatively, this patient may have a different disease because he was born to consanguineous parents, suggesting a possible recessive inheritance.
Although we did not perform a systematic review of all MR studies performed at our institute, to our knowledge no other patients have presented with the AOAD features here described. In our series, patients younger than 40 years were found to have periventricular WM abnormalities and postcontrast enhancement more frequently than patients older than 40 years. These findings might be related to decreased amounts and more limited distribution of Rosenthal fibers with increase in age of onset. Rosenthal fibers are more abundant in perivascular, subependymal, and subpial astrocytes. In the perivascular astrocytes, Rosenthal fibers accumulate in the endfeet that abut the endothelial cells, thus having a probable role in the regulation of the blood-brain barrier; malfunction of the barrier may explain the postcontrast enhancement that characterizes AD, particularly in young patients.2,15
The subpial prevalence of Rosenthal fibers may explain the peculiar distribution of signal intensity abnormalities observed in the upper brain stem in a few of our patients. The very thin, superficial, hyperintense rim seen on FLAIR images (Fig 2D) was not appreciated on T2-weighted images, probably because of overshadowing by the hyperintensity of the adjacent CSF.
The extension and conspicuousness of the MR and pathologic brain involvement in infantile AD progressively reduces in juvenile-onset and adult-onset cases, becoming limited to the lower brain stem and cervical spinal cord junction. In a similar fashion, the severity of the disease progressively diminishes, albeit the medullary location may also affect the vital functions. Why the adult MR pattern involves a very restricted area at the brain stem-spinal cord junction is unknown. A similar progressive limitation of brain areas involved in patients with adult-onset AD is also observed in other leukoencephalopathies (eg, in globoid-cell leukodystrophy or Krabbe disease).28
The explanation for the different phenotypes observed in infants and adults may reside in the functional effect of the different mutations that occur in the GFAP gene and likely depends on the site and type of the amino acid change.29 On the other hand, some mutations may cause both early-onset and late-onset AD or may show reduced penetrance in healthy parents, suggesting that modifier genes, intragenic effects, or environmental factors may contribute to phenotypic variability.30
As far as advanced MR techniques are concerned, data are scanty. To our knowledge, no reports on diffusion-weighted imaging in AD have been published. In our cohort, diffusivity was increased in areas of abnormal WM in patients 3 and 6 and was unaltered in normal-appearing WM. Therefore, the contribution of diffusion-weighted imaging seems to be limited. Additional studies with diffusion tensor imaging are warranted.
MR spectroscopy studies in juvenile forms of AD revealed increased mIns and decreased NAA in WM.31,32 MIns has a high intracellular concentration in astrocytes and is widely accepted as a glial marker; therefore, its increase is consistent with gliosis. In our series, mIns was increased only in patient 3, who had the most severe WM abnormalities. The absence of significant changes in all other patients indicates that increased mIns is not a sensitive marker of AOAD.
Conclusions
Progressive atrophy of the medulla oblongata and upper cervical spinal cord, with hyperintensities on T2-weighted images, is highly suggestive of AOAD. Involvement of the hilum of the dentate nucleus is frequent. In patients younger than 40 years, supratentorial periventricular abnormalities in signal intensity and spotty areas of postcontrast enhancement are observed. They are rarely seen in older patients. Expression of the disease becomes progressively limited with increase in age of onset.
Awareness of this peculiar MR pattern allows identification of patients to be submitted to genetic testing for the GFAP gene. Diagnosis may be proposed even in asymptomatic subjects examined with MR imaging for unrelated reasons and implies proper genetic counseling to families.
After GFAP testing was introduced, the number of AOAD diagnoses has progressively increased, and the adult-onset form of the disease may turn out to be more common than the well-known infantile one.
Acknowledgments
We thank Dr. Alberto Bizzi for reading the manuscript and for his helpful advice. We also thank the neurologists of our institute who took care of the patients included in the study.
Footnotes
Paper presented previously at: Annual Meeting of the American Academy of Neurology, April 28–May 5, 2007; Boston, Mass.
References
- Received December 18, 2007.
- Accepted after revision January 28, 2008.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Teaching NeuroImages: Neuroimaging in Adult-Onset Alexander Disease
- Practical approach to the diagnosis of adult-onset leukodystrophies: an updated guide in the genomic era
- Spinal cord involvement in adult-onset metabolic and genetic diseases
- CSF and Blood Levels of GFAP in Alexander Disease
- Adult-onset Alexander's disease mimicking degenerative disease
- Characteristic abnormal signals in medulla oblongata--"eye spot" sign: Four cases of elderly-onset Alexander disease
- A practical approach to diagnosing adult onset leukodystrophies
- Neuroimaging and clinical features in type II (late-onset) Alexander disease
- Reviewing the genetic causes of spastic-ataxias
- GFAP mutations, age at onset, and clinical subtypes in Alexander disease
- A case of sporadic adult Alexander disease presenting with acute onset, remission and relapse
- MR Imaging Characteristics and Neuropathology of the Spinal Cord in Adult-Onset Autosomal Dominant Leukodystrophy with Autonomic Symptoms