
Abstract
SUMMARY: Copper deficiency has been associated with a clinical syndrome, myeloneuropathy. Radiographic changes resembling B12 deficiency in the cervical spinal cord have been described. We present a case of copper deficiency myeloneuropathy, with cervical MR imaging findings resembling B12 deficiency, which partially reversed following copper supplementation. This is, to our knowledge, the first described case of radiographic improvement with copper supplementation.
Copper deficiency is an increasingly reported but under-recognized cause of neurologic dysfunction. This potentially treatable disorder manifests clinically as a profound sensory ataxia and can be associated with signal-intensity change in the dorsal columns of the spinal cord, resembling B12 deficiency. We present a patient with copper deficiency myeloneuropathy who had partial reversal of the MR imaging signal-intensity changes in the dorsal columns after copper replacement therapy.
Case Report
A 53-year-old woman was referred for neurologic evaluation of peripheral neuropathy. One year before our evaluation, she had been diagnosed with a myelodysplastic syndrome. Seven months before our evaluation, she developed lower limb numbness and gait unsteadiness, with numbness later affecting her upper limbs. These symptoms rapidly worsened over several months, and she lost the ability to ambulate and stand independently. Cognition and bowel and bladder function remained normal. The patient received a course of chemotherapy for her myelodysplastic syndrome but had not been treated for her “neuropathy.” The chemotherapy did not help the anemia. The patient had not been on zinc supplementation and had no antecedent history of malabsorption.
The patient’s speech, language, and mental status were normal. The findings of her cranial nerve examination were normal. She was moderately weak in distal upper and lower limb muscles, with milder weakness in her proximal limb muscles. Deep tendon reflexes were brisk in the upper and lower limbs. Hoffmann’s sign was noted in the upper limbs, but plantar responses were flexor. Complete absence of vibration sense was noted in the lower limbs, and joint position sense was absent in the lower limbs to the level of the hips. The patient was unable to stand without assistance, and she was unable to ambulate because of her severe proprioceptive deficits.
Diagnostic testing revealed anemia and leucopenia, without thrombocytopenia. Vitamin B12, folate, and methylmalonic acid levels were all normal. Other infectious and inflammatory serologic markers were negative for inflammation, and findings of a spinal fluid examination were normal. Serum copper and ceruloplasmin levels were markedly decreased at 0.05 ng/mL (0.75–1.45 ng/mL) and 1.37 g/mL (22.9–43.1 g/mL), respectively. Serum zinc levels were elevated at 2.50 (0.66–1.10). The patient’s electromyogram showed a mild polyneuropathy.
Tibial and median somatosensory-evoked potentials showed a combination of central and peripheral slowing in somatosensory pathways serving the upper and lower limbs.
Imaging and Clinical Follow-Up
Sagittal and axial fast spin-echo T2-weighted images of the cervical spine (Fig 1A, -B, respectively) demonstrated diffuse increase in signal intensity involving the dorsal columns of the cervical spinal cord extending from C1 to C7. Findings of sagittal T1-weighted images of the cervical spine were normal. Gadolinium was not given. The thoracic levels (not shown) were not involved. Findings of MR imaging of the brain were normal.
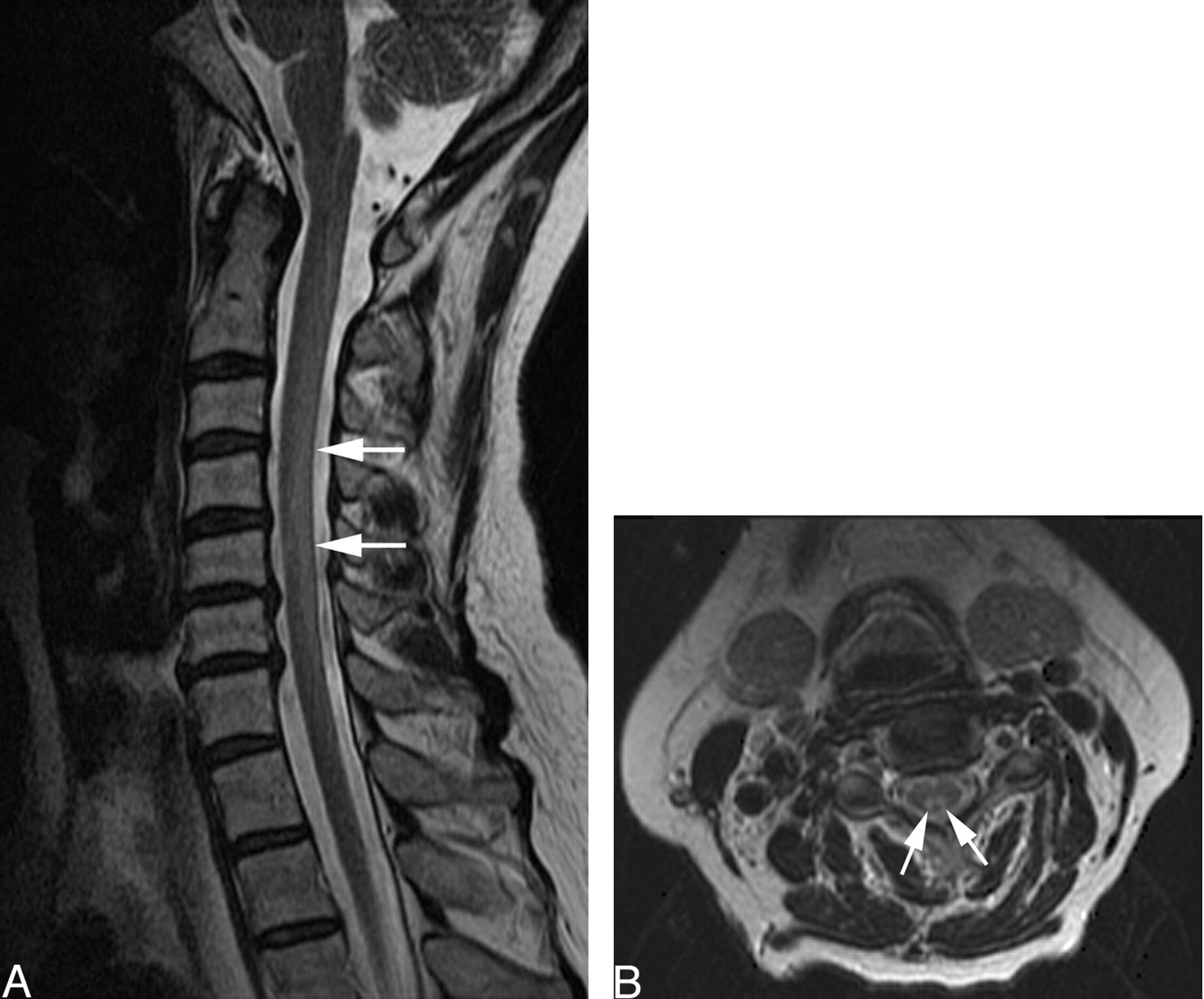
A and B, Sagittal (A) and axial (B) fast spin-echo images of the cervical spine before treatment demonstrate diffuse increase in signal intensity (arrows) involving the dorsal columns of the cervical spinal cord extending from C1 to C7.
The patient was treated with oral copper. Within 1 month of copper supplementation, the patient’s anemia resolved completely. When seen 4 months following the initiation of copper supplementation, the patient reported feeling stronger, though her sensory symptoms were unchanged. Neurologic examination showed resolution of the patient’s proximal weakness, though the neurologic examination was otherwise unchanged, notable primarily for a persistent severe sensory ataxia.
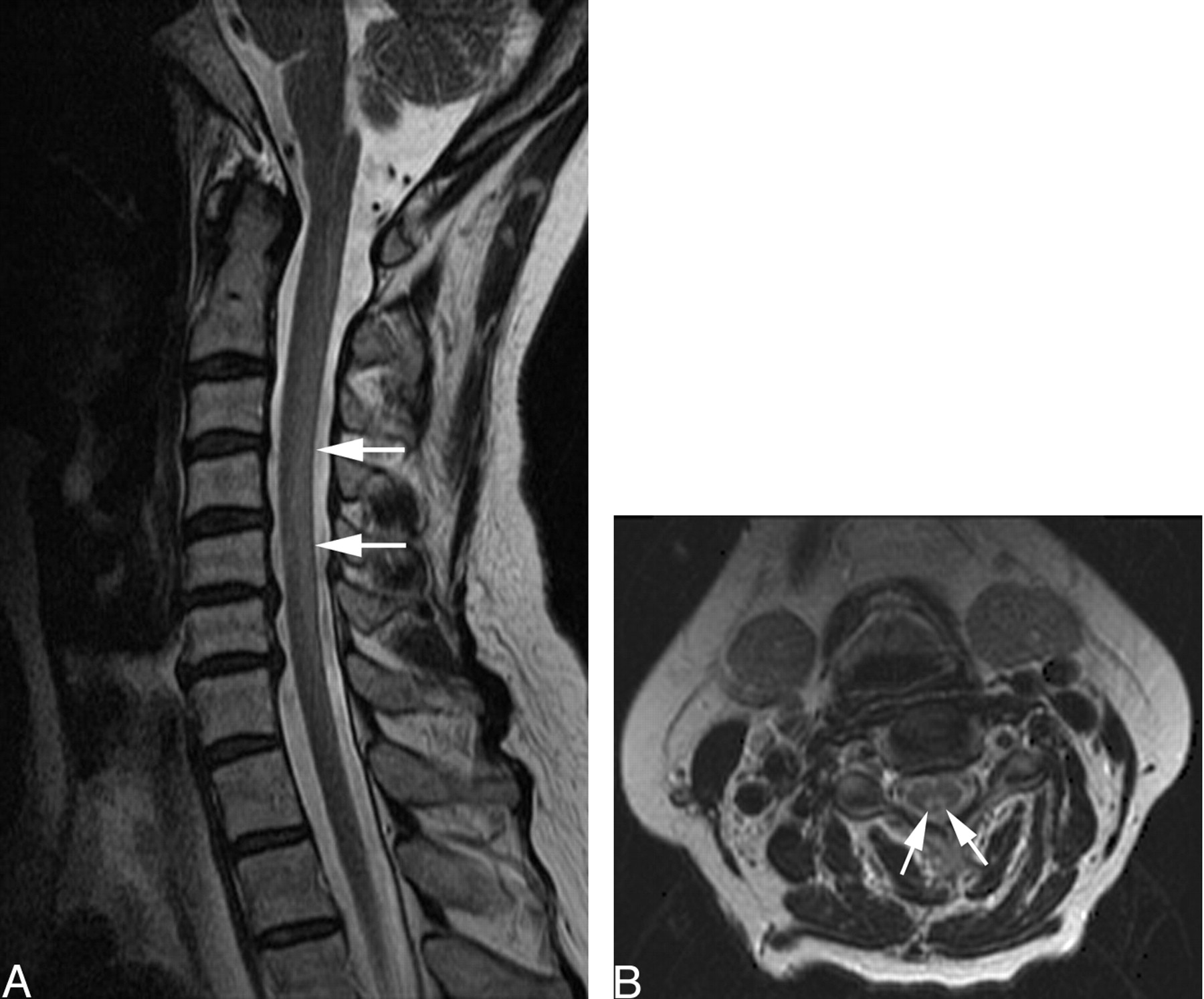
Follow-up cervical spine MR imaging examination 4 months later demonstrated marked improvement (Fig 2A, -B), with diminished T2 signal intensity in the dorsal columns.

A and B, T2-weighted sagittal (A) and axial (B) images after copper supplementation show marked resolution of signal intensity (arrows) in the cervical spinal cord.
Discussion
This patient presented with a refractory myelodysplastic syndrome and a severe, rapidly progressive sensory ataxia with laboratory evaluation revealing a severe copper deficiency, along with elevated serum zinc levels. There was clinical, electrophysiologic and radiographic evidence of a myeloneuropathy. Following copper supplementation, the myelodysplastic syndrome resolved, mild clinical improvement was noted, and near resolution of the cervical spine MR imaging abnormalities was seen.
Copper deficiency is an increasingly recognized cause of neurologic degeneration and is also an established cause of anemia and the myelodysplastic syndrome.1–8 Copper is an important cofactor in several enzymatic processes important in the function of the central nervous system, including cytochrome-c oxidase, copper-zinc superoxide dismutase, and dopamine β-hydroxylase. Known causes of copper deficiency include excess zinc ingestion, malabsorption, nephrotic syndrome, and parenteral nutrition (without adequate copper), and this condition can rarely be seen in malnourished infants.
Despite the recent recognition of the neurologic manifestations of acquired copper deficiency, there is a convincing precedent for this as a clinical syndrome. Menkes disease is an X-linked inherited disorder of copper malabsorption, which develops in infancy with various neurologic and systemic manifestations.9–11 Copper malabsorption in ruminant animals is a recognized cause of a progressive ataxic myelopathy, known as “swayback.” In “swayback,” necropsy has revealed demyelination in spinal cord white matter, which is similar pathologically to autopsied cases of Menkes disease.12 Copper supplementation in humans with acquired copper deficiency typically arrests further neurologic deterioration and can even result in clinical improvement.2 The myelodysplastic syndrome due to copper deficiency rapidly reverses with copper supplementation.
The mechanism of copper deficiency in this patient is not known. The patient denied exogenous excess zinc ingestion, had no prior history of gastric surgery, and did not have any other symptoms to suggest a more generalized malabsorption syndrome. In the absence of any other obvious mechanism, it is hypothesized that copper deficiency in such patients is likely due to a luminal copper transport or trafficking defect.1 Hyperzincemia was evident in our patient and has been reported in other cases of copper deficiency. In the absence of excess zinc ingestion, this has been postulated to be due to impaired luminal transport or trafficking of copper and zinc. Although zinc excess has been hypothesized by some to be the primary metabolic defect,13 several observations would argue otherwise. Inherited hyperzincemia has been described, without any clinical manifestations.14 Furthermore, excess zinc is known to stimulate the synthesis of metallothionein in intestinal enterocytes, to which copper binds with greater affinity than zinc.1 Copper subsequently is sloughed off into the intestinal lumen (and not absorbed).
The spinal MR imaging in this patient resembled that of subacute combined degeneration of the cord as is seen with vitamin B12 deficiency.15,16 Distinction from vitamin B12 deficiency based on imaging is not possible, though serum vitamin B12 and methylmalonic acid levels were normal in this case. The reason for the preferential involvement of cervical spine dorsal columns is unclear. The radiographic findings, however, are consistent with the profound proprioceptive loss seen clinically. There are previous reports of signal intensity change in the dorsal spinal cord in patients with copper deficiency.1,3,5 Kumar et al1 reported increase in T2 signal intensity in the dorsal columns in 3/13 patients with copper deficiency myelopathy. Our patient demonstrated improvement in the T2 signal-intensity change in the dorsal columns with time due to copper replacement therapy.
Brain MR imaging changes suggestive of demyelination in 2 other patients with copper deficiency have been reported,6 as well as more focal signal intensity change at midthoracic spinal cord segments in another patient with copper deficiency.7 Other than subacute combined degeneration of the cord, the differential diagnostic possibilities for abnormal increased T2 signal intensity in the dorsal cord include demyelinating disorders (eg, multiple sclerosis), infection (HIV vacuolar myelopathy and herpes virus infections), and ischemia. Demyelination such as that seen in multiple sclerosis or acute disseminated encephalomyelitis often does not involve segments greater than 2 vertebral bodies in length, whereas in our patient, there was continuous involvement of the dorsal cord spanning 7 vertebral bodies. Other inflammatory causes (HIV, herpes) are characterized by expansion of the cord and enhancement. The clinical presentation and electrophysiologic findings of our patient were not consistent with spinal cord ischemia, and otherwise normal serologic and spinal fluid results provide strong arguments against an occult infectious or inflammatory disorder.
References
- Received November 11, 2005.
- Accepted after revision November 23, 2005.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}