
Abstract
Summary: We present a case of a reversible spinal venous hypertensive myelopathy that occurred following a traumatic mediastinal hematoma. The mediastinal hematoma caused compression of the brachiocephalic vein, resulting in elevation of the venous pressures that ultimately resulted in dilation of the epidural venous plexus and spinal cord edema. The secondary neurologic deficits were the culmination of venous outflow obstruction at the level of the spinal cord that resolved on the resolution of the mediastinal hematoma.
Venous hypertensive myelopathy is the proposed mechanism leading to acute or subacute deterioration of neurologic function, mainly described in patients with spinal dural arteriovenous fistulas or arteriovenous malformations. Venous congestion is considered to be the major cause of progressive myelopathy (1–3). We present a case of reversible traumatic spinal venous hypertensive myelopathy that supports this theory.
Case Reports
A 13-year-old boy lost control while driving a go-cart and hit a mobile home at a high speed. At the scene, emergency personnel noted that he was awake, alert, oriented, and able to move all extremities. While being transported to the local hospital, he became dyspneic and lost consciousness. The patient was intubated and bilateral thoracostomy tubes were placed. On presentation to our institution, the patient was pharmacologically paralyzed and sedated. He became increasingly difficult to ventilate. An outside axial CT of the chest demonstrated a mediastinal hematoma, but no definite aortic arch injury. Extensive pneumomediastinum proved to be the result of a bronchopleural fistula at emergent bronchoscopy. A contrast-enhanced early arterial phase CT angiogram of the chest, obtained to better assess the aorta, demonstrated a right brachiocephalic artery dissection and pseudoaneurysm as well as a flow-limiting high-grade constriction of the left brachiocephalic vein by a mediastinal hematoma (Fig 1). The left upper extremity and head and neck venous drainage was diverted from the left brachiocephalic vein by reflux and distention of the accessory hemiazygos vein and the hemiazygos vein, which eventually drained to the heart via the inferior vena cava by multiple collaterals both above and below the diaphragm. One group of collaterals that were markedly distended were the internal epidural venous plexus of the lower thoracic spine.

CT angiogram of the chest at admission.
A, The left brachiocephalic vein (arrow) is compressed by a brachiocephalic artery dissection (not shown) and associated mediastinal hematoma (black arrowheads). An endotracheal tube, enteric tube, right thoracostomy tube (white arrowheads), right pneumothorax, and pneumomediastinum (asterisks) are present.
B, Note dilation of the accessory hemiazygos vein (white arrowhead) and enlarged vein within the neural foramen (black arrowhead).
C, Maximum-intensity-projection image of the CT angiogram demonstrates constriction of the brachiocephalic vein (arrowhead) and dilation of the accessory hemiazygos vein (large arrow) and epidural venous plexus (small arrows).
The propofol (Diprivan, AstraZeneca, Newark, DE) and vecuronium (Norcuron, Oragon USA Inc., Roseland, NJ) were discontinued during the following week as the patient’s status stabilized. At this time, it was noted on physical examination that the patient had bilateral lower extremity paralysis and decreased reflexes. He also had decreased sensation below the T12 level. MR imaging of the spine demonstrated thoracic cord edema with prominent tubular flow voids posterior to the spinal cord (Fig 2). There was no evidence of spinal fractures, hemorrhage within the cord, or cord compression. Postcontrast MR imaging was not performed. With venous hypertension as our leading diagnosis and an angiogram that demonstrated a stable brachiocephalic artery pseudoaneurysm, we decided to monitor the improvement of the mediastinal hematoma. As the venous obstruction resolved, so did the patient’s neurologic deficits (Fig 3). Follow-up MR imaging of the spine was not performed. The patient’s lower extremity paralysis resolved during the course of 3 weeks with daily occupational and physical therapy.

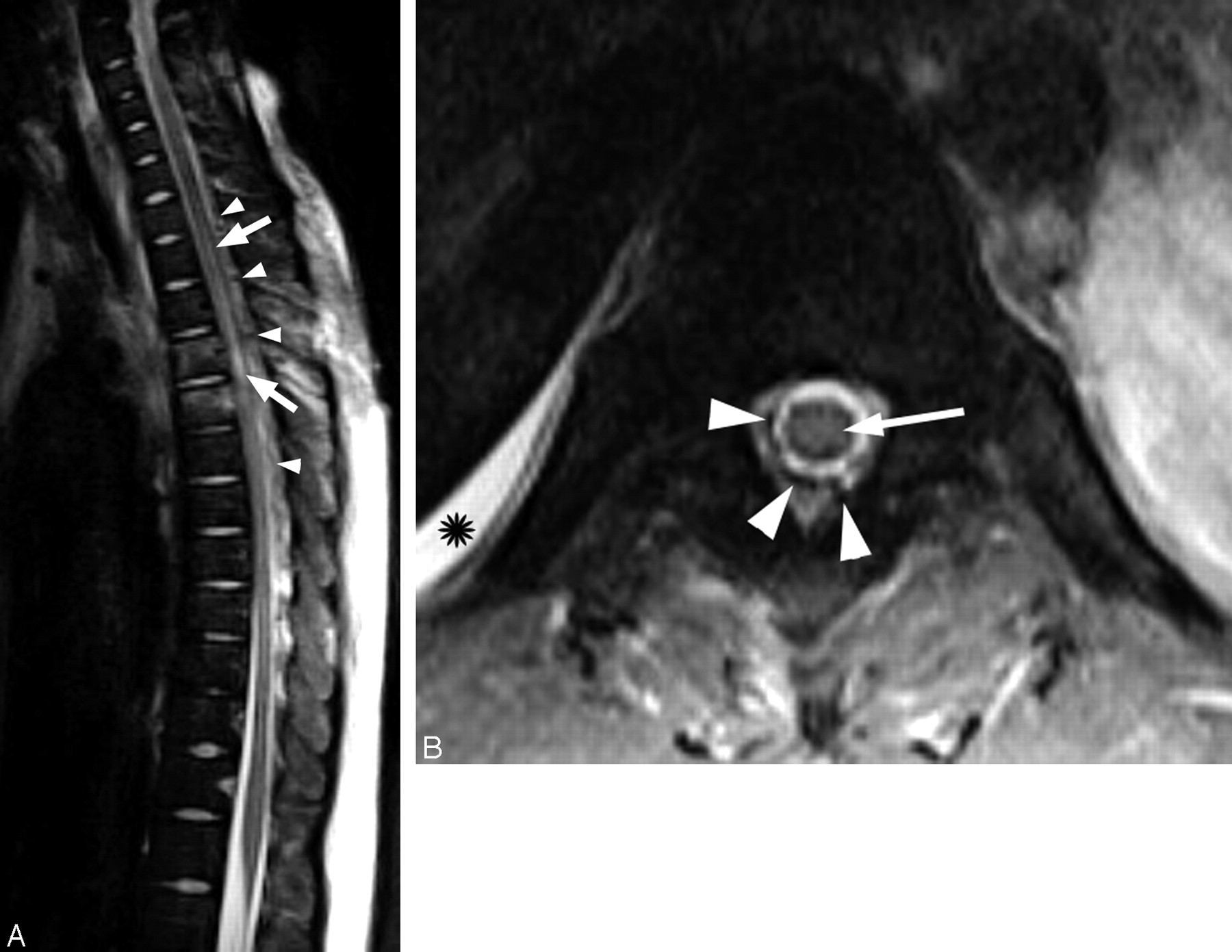
Thoracic spine MR imaging.
A, Sagittal T2-weighted MR image of the thoracic spine shows increased signal intensity within the spinal cord (arrows). There are prominent low-signal-intensity structures (arrowheads) along the posterior surface of the cord that represent flow voids from the dilated perivertebral plexus. The increased signal intensity of the T3 and T4 vertebral bodies is consistent with bone contusions, without loss of vertebral body height.
B, Axial T2-weighted MR image of the spine demonstrates tubular flow voids in the epidural space (arrowheads) that represent the dilated epidural venous plexus. There is increased signal intensity within the central portion of the spinal cord (arrow) compatible with edema. Pleural effusion is also noted on the right (asterisk).

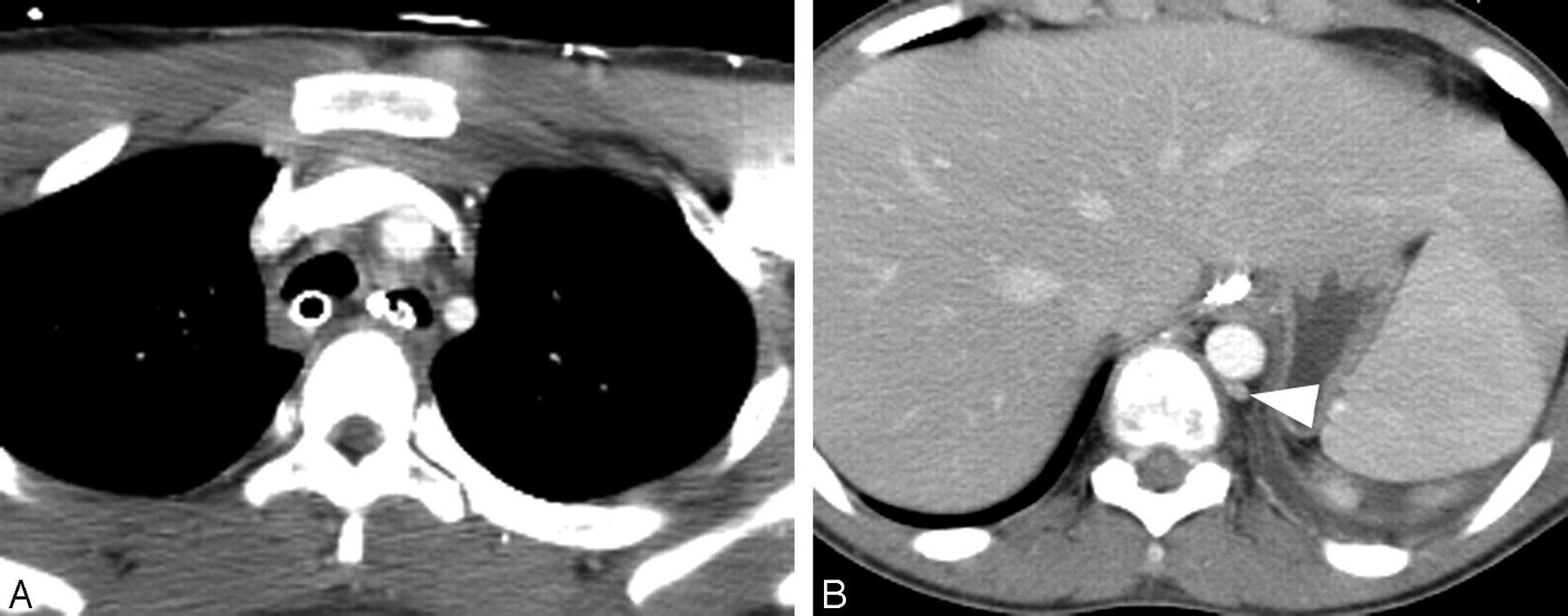
Follow-up CT angiogram of the chest obtained 10 days after the initial CT angiogram.
A, Note interval resolution of the brachiocephalic vein compression.
B, Previously identified dilated accessory hemiazygos vein (arrowhead) and epidural venous plexus have resolved.
Discussion
Clinically, venous hypertensive myelopathy presents with a progressive myelopathy that may be reversible with the appropriate management. The inference is that impaired venous drainage results from a decrease in the arteriovenous pressure gradient at the spinal cord, leading to increased intramedullary pressure and congestion. This sequence of events leads to spinal cord edema and a concomitant decrease in perfusion, resulting in ischemia and hypoxia (2, 4, 5). This theory is predicated on the fact that histologic changes, including pial venous engorgement, cord edema, and ischemia/infarction have been demonstrated in patients presenting with progressive hypertensive myelopathy.
Also, surgical series have shown that relief of the outflow obstruction halts and even reverses the neurologic deficits (5). The understanding of this reversible myelopathy is based on literature concerning the evaluation and consequences of dural arteriovenous fistulas and arteriovenous malformations at the level of the spinal cord. If, as conjectured, the myelopathy is related to the venous hypertension and not, as other theories proposed, to thrombosis or vascular steal, then a downstream venous obstruction should cause similar clinical, radiologic, and histologic findings. Evidence of macroscopic obstruction at a site remote from the spine would not only add validity to the hypothesis of venous hypertensive myelopathy, but also might possibly alter the way we evaluate and treat patients with an acute myelopathy.
The patient in this case did not demonstrate any neurologic deficits within the first few hours after the accident. The myelopathy became apparent only after approximately 1 week, when the paralytic medications were withdrawn. This temporal relationship coincided directly with the progressive dilation of the venous system documented on serial CT examinations. This coincidence implies a subacute onset of the neurologic deficits. Furthermore, the neurologic symptoms resolved with time as the venous obstruction abated. We believe these findings mirror similar results in patients presenting with and treated for spinal dural arteriovenous fistulas and arteriovenous malformations related to presumed venous hypertension.
Radiologic signs of venous hypertension in association with spinal dural arteriovenous fistulas and arteriovenous malformations are well described. These include spinal cord swelling, increased T2 signal intensity within the cord, parenchymal enhancement, enlargement of the vessels along the cord surface, and possibly peripheral hypointensity of the cord on T2-weighted images (6–8). In our case, the MR imaging of the spine demonstrated spinal cord edema, a relatively nonspecific finding; however, there were prominent tubular flow voids along the dorsal surface of the spine. This finding, although less commonly seen, is very specific for spinal venous hypertensive myelopathy (7).
We present an example of trauma-related venous hypertension resulting in secondary spinal cord injury. A recent case report further corroborates this cause and effect by describing venous hypertensive myelopathy related to obstruction from disk herniation (9). We believe that these secondary insults on the spinal cord further validate the presumed pathophysiology of spinal venous hypertensive myelopathy. These cases prove that it is particularly important to evaluate for this entity in all patients presenting with an acute or subacute myelopathy.
References
- Received August 31, 2004.
- Accepted after revision October 11, 2004.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}