
Transsynaptic injury caused by excitotoxic amines is a specific type of injury in the peripheral and central nervous systems. Recent studies have shown that the receptors related to excitotoxic mechanisms are widely distributed in the brain, not only in the gray matter (neurons and astrocytes) but also in the white matter (astrocytes, oligodendrocytes, myelin sheaths, and axons) (1).
The object of this review is to illustrate and describe excitotoxic mechanisms in various acute neurologic conditions. Such conditions include infarction, hypoxic ischemic encephalopathy (HIE), the early phase of wallerian and transneuronal degeneration, shaken baby syndrome, status epilepticus, a corpus callosum lesion related to either seizures or antiepileptic drugs, diffuse axonal injury, toxic or metabolic leukoencephalopathy, the acute phase of multiple sclerosis, and Creutzfeldt-Jakob disease (CJD). Excitotoxic brain injury is considered a final common pathway for various neuropathologic conditions and causes cytotoxic edema. Diffusion-weighted (DW) imaging is especially useful for the early detection of cytotoxic edema as an area of abnormal hyperintensity associated with a decreased apparent diffusion coefficient (ADC).
Excitotoxic Mechanisms
Glutamate, aspartate, and glycine are the dominant excitatory amino acids and the primary neurotransmitters in about one-half of all the synapses in the brain (2). Among them, glutamate is the most important and responsible for many neurologic functions including cognition, memory, movement, and sensation. In pathologic conditions, glutamate mediates neuronal injury or neuronal death, particularly through activation of the N-methyl-d-aspartate (NMDA) receptor subtype of the glutamate receptor.
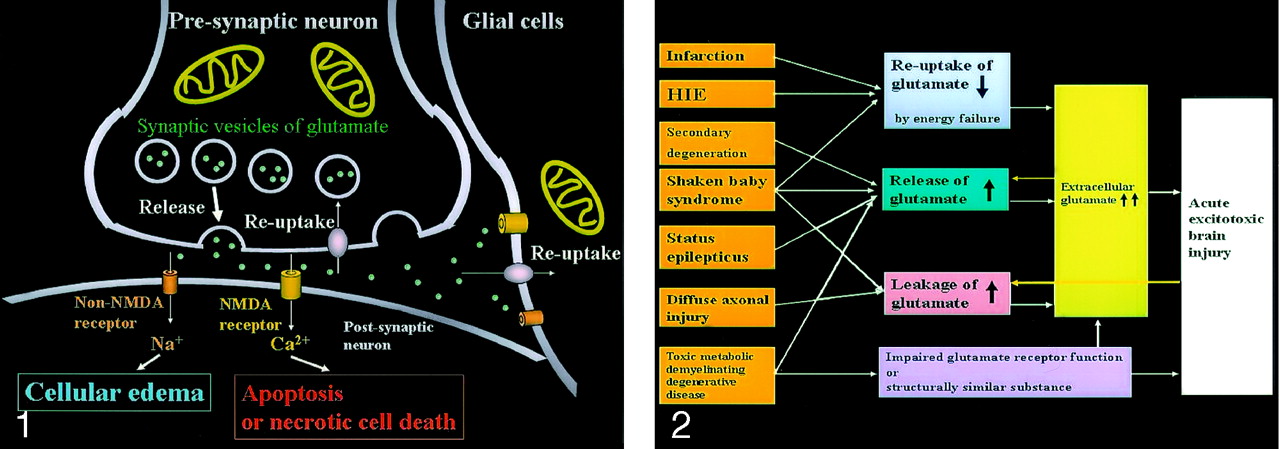
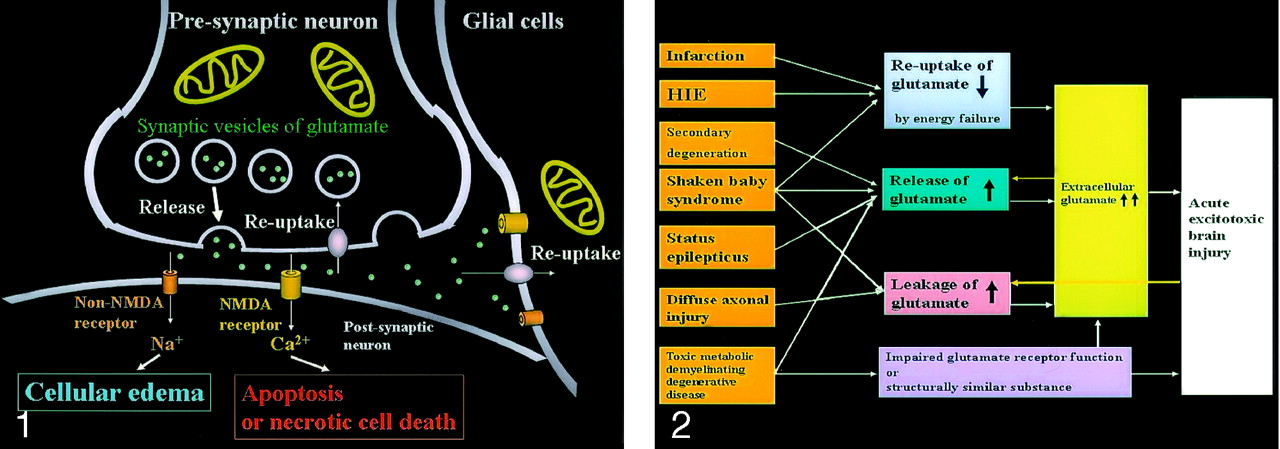
Neuronal glutamate is released from the presynaptic terminal of neuronal axons into the synaptic cleft and then works as a neurotransmitter (Fig 1). Reuptake of extracellular glutamate takes place at the presynaptic terminals and adjacent glial cells. Mitochondria provide energy for the reuptake of glutamate. The excessive glutamate binding to NMDA receptors allows entry of Ca2+ into the postsynaptic neuron, causing necrotic cell death or apoptosis, whereas the excessive glutamate binding to non-NMDA receptors allows entry of Na+ into the postsynaptic neuron, resulting in cytotoxic edema. Because glial cells also have these receptors, the excessive glutamate leads to glial cell swelling, which seems to protect the neurons from excitotoxic injury.
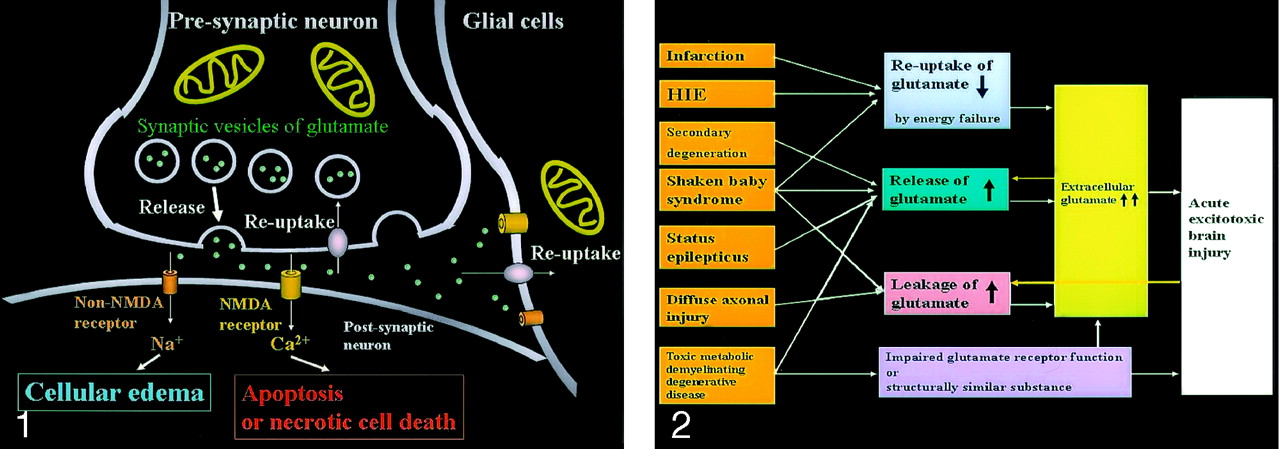
Excitotoxic mechanisms in the neuron glial unit. Glutamate is released from the presynaptic neuron into the synaptic cleft. It then undergoes reuptake into the presynaptic neuron and adjacent glial cells. Mitochondria provide energy for the reuptake. Excessive glutamate binding to non-NMDA receptors allows entry of Na+ into the postsynaptic neuron, resulting in cytotoxic edema; excessive glutamate binding to NMDA receptors allows entry of Ca2+ into the postsynaptic neuron, causing necrotic cell death or apoptosis (From Moritani T, Ekholm S, Westesson PL. Diffusion-Weighted MR Imaging of the Brain. Berlin: Springer-Verlag; 2004. Used with permission.)
Excitotoxic mechanisms play an important role in cellular damage in various diseases, and glutamate excitotoxicity is the final common pathway resulting in brain injury for many seemingly unrelated diseases. Increased extracellular glutamate is a direct cause of excitotoxic brain injury (Fig 2). In acute excitotoxic injury, excessive extracellular glutamate results from 1) decreased reuptake energy failure, 2) increased release by excessive depolarization of neuronal membranes or by intracellular accumulation, and 3) leakage due to disruption of axonal membranes. Acute excitotoxic injury can also be related to functional failure of the glutamate receptor and the presence of substances structurally similar to glutamate, such as hydroxyglutarate or glutarate, at the receptor sites. Excessive extracellular glutamate depolarizes injured adjacent glial cells or neurons and in turn causes release or leakage of glutamate. This mechanism is self-propagating through neuron-glial cell units and through transaxonal or transynaptic routes along the white matter fiber tracts.

Acute excitotoxic brain injury in various diseases associated with acute excitotoxic brain injury. Mechanisms are classified by 1) decreased reuptake; 2) increased release; 3) leakage due to disruption of axonal membranes; and 4) others, including impaired glutamate receptor function or a substance structurally similar to glutamate. Combinations of mechanisms vary according to each disease process. Two positive feedback loops (yellow arrow) are present: 1) Increased extracellular glutamate depolarizes adjacent neurons that release intracellular glutamate, and 2) neuronal injury causes leakage of glutamate. These make this mechanism self-propagating via neuron-glial cell units and via transaxonal or transynaptic routes along the fiber tracts.
Brain Infarction
During ischemia, cytotoxic edema occurs primarily because of energy failure in neurons and astrocytes. It may extend into the region of the penumbra by means of neuron-glial cell units and synapses via excitotoxic mechanisms (3). Either energy failure or excitotoxic mechanisms disable the sodium-potassium pump, allowing extracellular sodium and water to enter the cell. Elevation of intracellular calcium ion levels may trigger protease and lipase production resulting in infarction, gliosis, or delayed neuronal loss. In experimental studies, NMDA-type glutamate receptor antagonists (MK-801) reduce the volume of ischemic injury after occlusion of the middle cerebral artery (MCA) (4). This finding indicates that the pathophysiologic findings of ischemic penumbra are associated with excitotoxic injury related to glutamate.
Diffusion-weighted imaging shows cytotoxic edema of ischemic lesions as hyperintense with decreased ADC, which may extend into the ischemic penumbra as a result of its propagation by excitotoxic mechanisms (Fig 3).

Hyperacute infarction (2 hours after onset) in a 39-year-old man with the left internal carotid artery dissection presenting with right-sided weakness. (From Moritani T, Ekholm S, Westesson PL. Diffusion-Weighted MR Imaging of the Brain. Berlin: Springer-Verlag; 2004. Used with permission.)
A, Fluid-attenuated inversion recovery (FLAIR) image shows no apparent parenchymal abnormalities, but linear hyperintensities represent slow flow in the peripheral vessels (arrows). B and C, DW images show a mildly hyperintense lesion with decreased ADC in the left frontoparietal white matter; this represents cytotoxic edema extending into the ischemic penumbra, propagated by excitotoxic mechanisms (arrows).
D, Perfusion-weighted image shows prolonged mean time to peak in the entire left MCA and anterior cerebral artery territories.
Wallerian Degeneration and Transneuronal Degeneration
Cerebral infarction in the MCA territory can cause wallerian degeneration of the corticospinal tract and transneuronal degeneration in the ipsilateral substantia nigra. Wallerian degeneration is antegrade degeneration of axons and myelin sheaths resulting from injury of the proximal axons or cell bodies. Transneuronal degeneration in the substantia nigra occurs secondary to striatal infarction (5). Cytotoxic edema occurring in the early phase of degeneration may result from excitotoxic mechanisms propagating through axons or synapses (6).
DW imaging shows the early phase of wallerian and transneuronal degeneration as hyperintense with decreased ADC, which presumably represents axonal, intramyelinic or astrocytic swelling (7–10) (Fig 4).

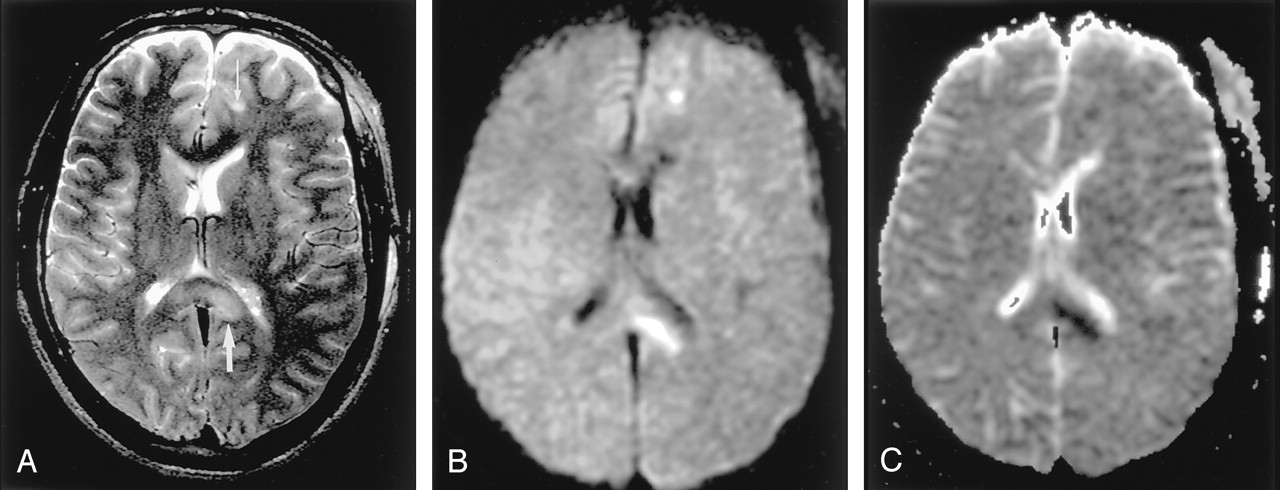
Wallerian degeneration in a 76-year-old man with a large right MCA infarct (6 days after onset).
A, T2-weighted image shows a hyperintense right MCA infarct, including the left putamen and caudate head.
B and C, DW images at the level of the midbrain reveal hyperintense lesions with decreased ADC, not only in the right MCA are, but also in the right cerebral peduncle (arrow) including the substantia nigra (arrowhead). This finding is presumably due to the excitotoxic injury propagated through transaxonal and transynaptic routes along the fiber tracts.
Hypoxic Ischemic Encephalopathy
In HIE, energy depletion in neurons and glial cells causes decreased reuptake of glutamate, which leads to increased extracellular glutamate. The distribution of HIE is different in adults and children. During the perinatal period, the developing brain is particularly vulnerable to excitotoxic injury. NMDA receptors dominate in the immature brain when synaptic transmission is inefficient and extremely plastic (2). The high rate of myelination and formation of synapses (synaptogenesis) can result in vulnerability from ischemic injury.
The predilection of the lesions for the putamen, thalamus, and peri-Rolandic cerebral cortex in profound HIE coincide with areas of vulnerability of caused by energy failure. One potentially important link among these areas is their interconnection by excitatory circuits (11). Therefore, overactivity at one location in these excitatory pathways could propagate to other locations through their synaptic connections. The internal capsule, cerebral peduncle, and corpus callosum can be secondarily involved through these pathways; this is also known as wallerian and transneuronal degeneration.
DW imaging clearly depicts lesions in the basal ganglia and white matter and along the corticospinal tract and corpus callosum when conventional CT and MR images are normal, or they may show only subtle abnormalities in these areas (Figs 5 and 6) (12).

Neonatal HIE in a 6-day-old boy with profound perinatal asphyxia.
A, On the T2-weighted image, gray matter–white matter delineation is partially obliterated. The anterior and posterior aspects of the corpus callosum show high signal intensity.
B and C, DW images show diffuse hyperintensity with decreased ADC in the corpus callosum (arrows), internal capsules, thalami, and white matter. This distribution may be related to excitatory circuits. The neonatal brain seems to be highly vulnerable to acute excitotoxic injury.
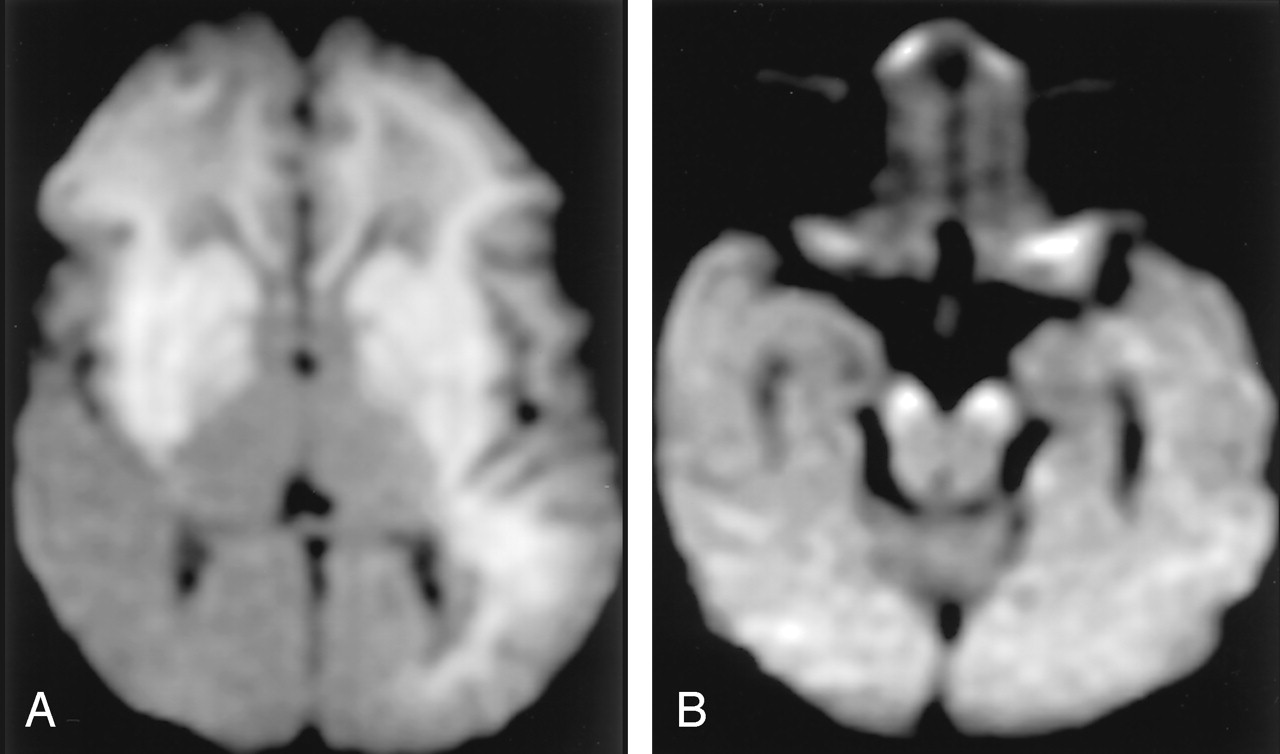
Neonatal HIE in a 10-day-old boy with profound perinatal asphyxia.
A, DW image shows extensive hyperintense lesions involving the frontotemporoparietal white matter, internal capsules, and basal ganglia.
B, DW image at the level of the midbrain shows hyperintense lesions with decreased ADC (not shown) in the bilateral cerebral peduncles. These findings represent the early phase of wallerian degeneration.
Shaken Baby Syndrome
With shaken baby syndrome, glutamate levels in the CSF are extremely high (13). For example, in experimental acute subdural hematomas in shaken infant rat brain, glutamate increases more than seven times over baseline levels (14). Although the pathogenesis of brain parenchymal injuries remains unknown, it seems closely related to combined excitotoxic mechanisms: increased release and leakage of glutamate with traumatic stimuli or shearing injury and decreased reuptake of glutamate with hypoxia or ischemia. Usually, the distribution of widespread parenchymal injury is neither related to the vascular territories nor the location or size of acute subdural hematomas on CT and MR imaging. Widespread parenchymal injury may be related to the distribution of NMDA receptors in neonates and infants. A unique predilection to shearing injury may also be important.
DW imaging is useful in detecting cytotoxic edema due to excitotoxic brain injury (Fig 7). The severity of abnormalities on DW images is correlated with clinical outcome (15–17). Neuroprotective effects mediated by several kinds of selective glutamate receptor antagonists, such as MK-801, dextromethorphan, or tirilazad, are reported in animal studies (4, 18–20).

Shaken baby syndrome in a 2-year-old girl.
A, T2-weighted image shows bilateral hyperintense lesions in the frontal and temporo-occipital lobes, basal ganglia, and corpus callosum. Subdural hematomas are seen as linear, hypointense lesions along the interhemispheric fissure (arrows).
B, T1-weighted image shows subdural hematomas in the posterior fossa in addition to the interhemispheric fissure (arrows).
C and D, DW images show the brain parenchymal injury as hyperintense with decreased ADC that represents cytotoxic edema presumably due to injuries with combined excitotoxic mechanisms.
Diffuse Axonal Injury
Diffuse axonal injury is considered to be caused by excitotoxic mechanisms, particularly those involving glutamate and NMDA receptors (21). Axonal damage often occurs at the node of Ranvier, a short interval between the myelin sheaths (processes of oligodendrocytes), resulting in a traumatic defect in the axonal membrane; this defect allows the leakage of glutamate into the extracellular space (22). The astrocytic end-foot is located on the axon at the node of Ranvier and may protect the axons (Fig 8). Excessive extracellular glutamate leads to axonal swelling and cytotoxic edema of glial cells, which may contribute to diffusion abnormalities resulting in necrosis, axonal degeneration, and gliosis.

Diffuse axonal injury is presumably due to the leakage of glutamate at the node of Ranvier. The astrocytic end-foot is located on the axon at the node of Ranvier and may protect the axons.
DW imaging shows hyperintense lesions associated with decreased ADC. This is often present in the corpus callosum, fornix, gray matter–white matter junction, and brainstem (including cerebellar peduncles) (23) (Fig 9).
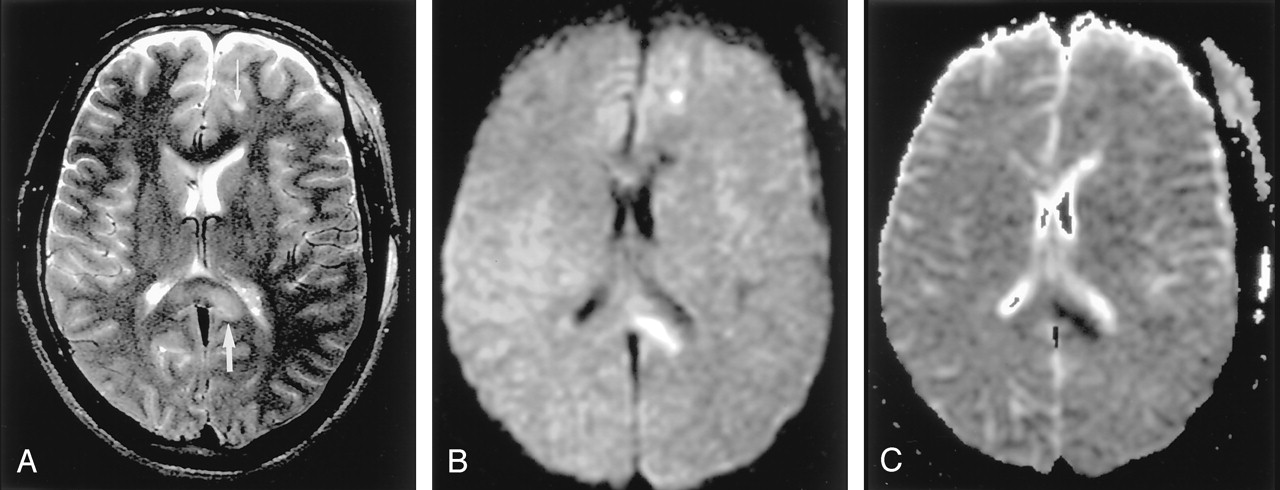
Diffuse axonal injury in a 20-year-old man after motor vehicle accident.
A, T2-weighted image shows hyperintense lesions in the splenium of the corpus callosum and left frontal gray matter–white matter junction (arrows).
B and C, DW images show these lesions as hyperintense with decreased ADC; they represent cytotoxic edema presumably due to the leakage of glutamate from the axon.
Herpes Encephalitis
In acute encephalitis, concentrations of glutamate and glycine in the CSF are significantly increased (24). This observation suggests that an excitotoxic mechanism plays a role in neuronal damage in herpes encephalitis. Excessive glutamate release, due to free radicals generated during the immune response to infections, might initiate the excitotoxicity.
The distribution of herpes encephalitis differs according to the age of the patient. In older children and adults, encephalitis due to herpes simplex type 1 usually involves the medial temporal lobes, inferior frontal lobes, and insula. DW imaging shows signal intensity abnormalities in these areas (Fig 10). Neonatal encephalitis due to herpes simplex type 2 usually affects the cerebral cortex and white matter in a more global fashion. Widespread brain lesions in neonatal herpes encephalopathy are presumably related to the vulnerability to excitatory amines in the neonatal brain (2). In neonatal herpes encephalitis, DW imaging shows widespread, asymmetric abnormalities in both hemispheres, including the basal ganglia and thalami (Fig 11).

Herpes encephalitis type 1 in a 48-year-old man presenting with headache and fever.
A, T2-weighted image shows hyperintense lesions in both medial temporal lobes, including the right hippocampus (arrows).
B, DW image clearly shows these bilateral lesions as hyperintense.
C, ADC maps show partially decreased ADC of these lesions (arrows).

Herpes encephalitis type 2 in a 2-week-old girl.
A, T2-weighted image shows bilateral hyperintense lesions in the basal ganglia, thalami, and frontotemporal regions.
B and C, DW images show asymmetric but extensive hyperintense lesions with decreased ADC in these areas. This extensive distribution of the lesions is presumably related to vulnerability of the developing brain to excitotoxic injury.
Status Epilepticus
In status epilepticus, neuronal injury results primarily from an excitotoxic mechanism mediated by intrinsic neuronal seizure activity (25). During status epilepticus, neuronal seizure activity increases the release of glutamate from the presynaptic terminals of neuronal axons. Excessive glutamate crosses the synaptic cleft to bind to NMDA and non-NMDA receptors, which causes cytotoxic edema in neurons and glial cells, leading to apoptosis or selective neuronal necrosis. Astrocytes play an important role in cellular and tissue repair by detoxifying excessive glutamate (26, 27). Cytotoxic edema of reactive astrocytes in the acute phase is presumed to be responsible for the reversible signal intensity abnormalities (28). Encephalopathy with status epilepticus often involves the hippocampus, other parts of the limbic system, the thalamus, and the cerebellum.
This distribution of lesions on DW imaging seems to be related to the distribution of NMDA-type glutamate receptors, which are concentrated in the hippocampus and in other parts of the limbic system (Fig 12).
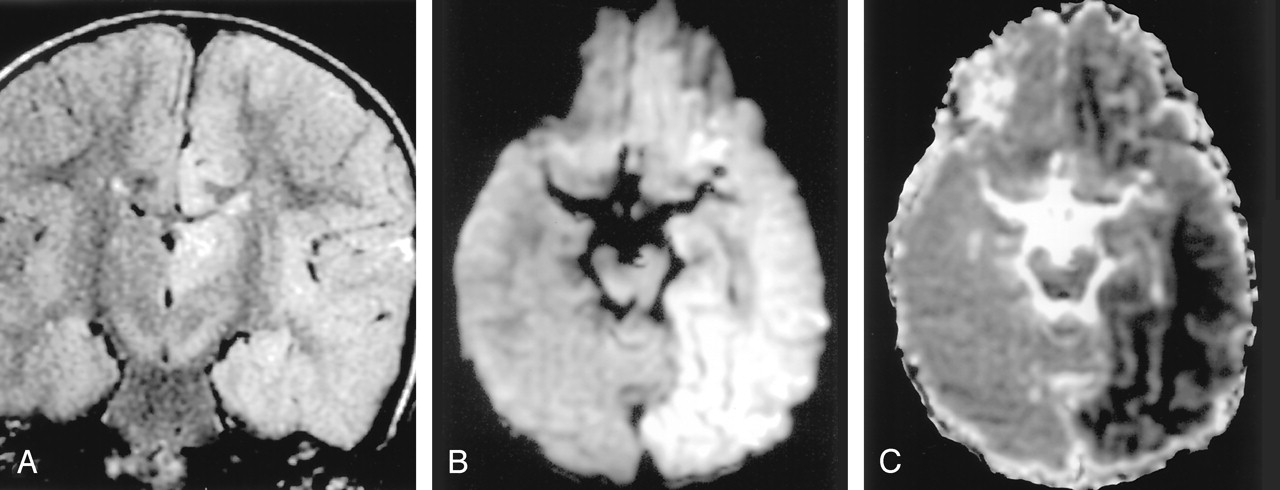
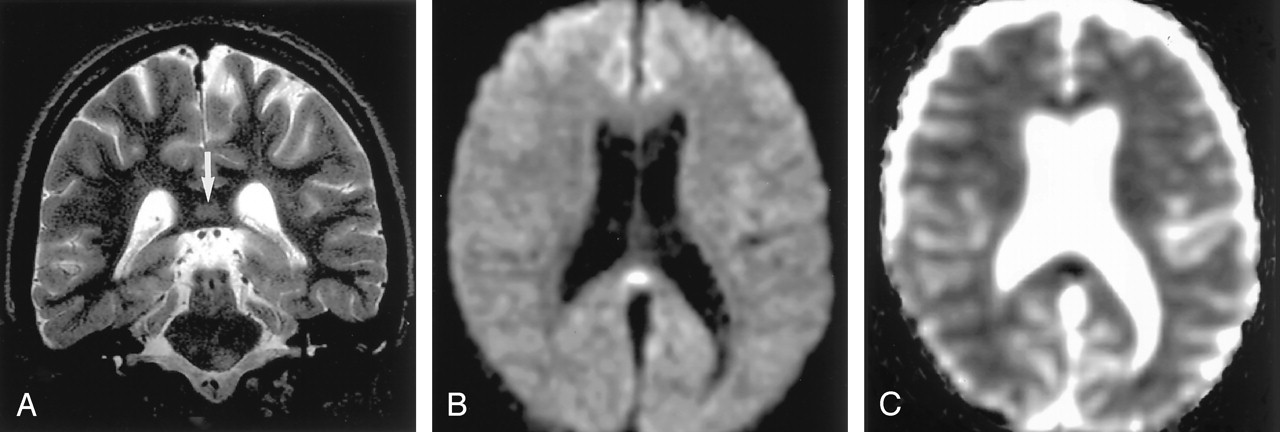
Status epilepticus in a 2-year-old girl.
A, Coronal FLAIR image shows diffuse hyperintense lesions in the left hemisphere including the left thalamus, cingulate gyrus, and hippocampus.
B and C, DW images show these lesions as hyperintense with decreased ADC. They represent cytotoxic edema due to excitotoxic injury mediated by neuronal seizure activity. These lesions were partially reversed on follow-up MR imaging (not shown).
Focal Lesion in the Splenium of the Corpus Callosum in Epilepsy
A unique focal area of diffusion abnormality has been observed in the splenium of the corpus callosum in the immediate postictal period. Causes of a focal lesion in the corpus callosum are speculated to be seizures, medications, or both (29–31). The transhemispheric connection of seizure activity may be the cause of transient focal edema. Seizure activity propagates through the splenial collosal fibers to the contralateral hemisphere. The splenium contains decussating fibers originating in the temporal lobes; these are likely to be the pathway for the intrahemispheric propagation of seizure activity originating in the temporal-lobe focus.
The transient focal edema in the splenium of the corpus callosum is also observed with the abrupt with- drawal or reduction of antiepileptic medications such as phenytoin, carbamazepine, and vigabatrin (30). This effect is mediated by the influence of antiepileptic medications on fluid-balance systems, namely the arginine-vasopressin system (31). Seizure activity or the effects of medications can lead to excitotoxic injury, resulting in reversible cytotoxic edema in astrocytes or myelin sheaths. Studies have demonstrated the presence of a large number of glutamate receptors and high enzymatic activity in the corpus callosum (1, 32).
Conventional T2-weighted MR imaging shows a nonhemorrhagic, hyperintense lesion in the splenium of the corpus callosum, which is slightly hypointense on T1-weighted images. DW imaging shows an acute lesion in the splenium as hyperintense with decreased ADC (Fig 13) (33).

Focal lesion in the splenium of the corpus callosum in a 9-year-old child with intractable partial seizures beginning at age 4.
A, T2-weighted image 3 days after a seizure shows a discrete, focal hyperintense lesion in the central portion of the splenium (arrow).
B and C, DW images show this lesion as hyperintense and associated with decreased ADC.
Osmotic Myelinolysis
Central pontine myelinolysis and extrapontine myelinolysis represent destruction of the myelin sheaths in characteristic places in the brainstem and cerebrum. Organic osmolytes, including glutamate, glutamine, betamine, or taurine, have been implicated in the pathogenesis of myelinolysis induced by the rapid correction of severe hyponatremia (34). Pathologic findings include destruction of myelin sheaths, although the nerve cells and axons are relatively spared.
DW imaging can depict the lesions in the early phase as hyperintense with decreased ADC, which represents cytotoxic edema (Fig 14).

Central pontine myelinolysis in a 14-year-old female adolescent. (From Moritani T, Ekholm S, Westesson PL. Diffusion-Weighted MR Imaging of the Brain. Berlin: Springer-Verlag; 2004. Used with permission.)
A, T2-weighted image shows a hyperintense lesion in the pons.
B and C, DW images show this lesion a hyperintense with mildly decreased ADC. This finding represents cytotoxic edema seen in the early phase of central pontine myelinolysis
Methotrexate-Induced Leukoencephalopathy
Intrathecal or intravenous methotrexate, either with or without radiation therapy, can occasionally cause diffuse white matter changes (35). Microvascular occlusion is considered one of the causes of white matter ischemic changes. Methotrexate itself is not toxic to astrocytes, neurons, or the neurite network. Another cause of neurotoxicity is thought to be mediated by the enzymatic release of glutamate from methotrexate. Glutamate excitotoxity can damage myelin sheaths and axons. NMDA receptor antagonists can protect against glutamate neurotoxicity (36).
MR imaging shows diffuse or multifocal white matter lesions that are hyperintense on T2-weighted images. DW imaging in the acute phase of the disease shows diffuse hyperintensity with decreased ADC in the white matter before conventional MR imaging is able to depict the lesions (Fig 15). Pathologically, the diffuse white matter lesion probably represents intramyelinic or axonal edema (Fig 15).

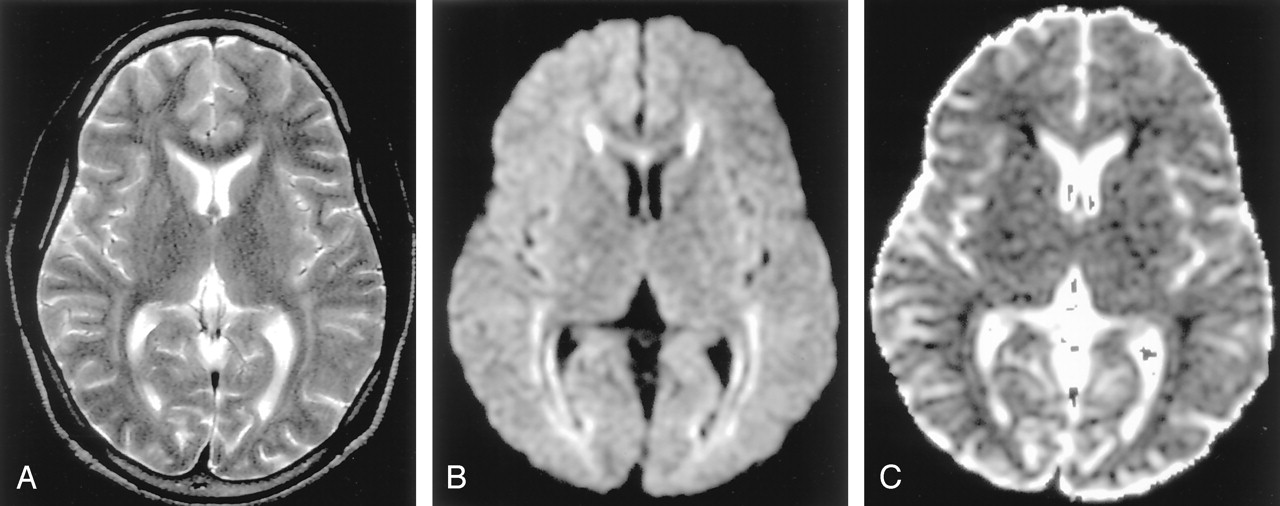
Methotrexate leukoencephalopathy in a 50-year-old woman. (From Moritani T, Ekholm S, Westesson PL. Diffusion-Weighted MR Imaging of the Brain. Berlin: Springer-Verlag; 2004. Used with permission.)
A, T2-weighted image is normal.
B, DW image shows diffuse hyperintensity in the bilateral corona radiata and extending into the central semiovale.
C, ADC map shows diffuse white matter lesions with decreased ADC, which represents pure cytotoxic edema presumably due to the enzymatic release of glutamate.
D, Pathologic specimen shows pure intramyelinic edema.
Phenylketonuria and Cerebral Organic Acid Disorders
Phenylketonuria is an autosomal recessive disorder caused by a deficiency of phenylalanine hydroxylase. l-phenylalanine impairs glutamate receptor function and thus contributes to brain dysfunction in phenylketonuria (37). Pathologic findings include delayed or defective myelination, intramyelinic edema, diffuse white matter vacuolation, demyelination, and gliosis (38).
MR imaging shows hyperintense lesions on T2-weighted images in the periventricular parietal and occipital regions, and in more severe cases, extending to the frontal and subcortical white matter (39). DW imaging shows these lesions as hyperintense with decreased ADC, probably the result of intramyelinic edema and astrocytic swelling, presumably due to excitotoxic injury (40) (Fig 16). With appropriate dietary control, these MR imaging abnormalities can completely resolve.
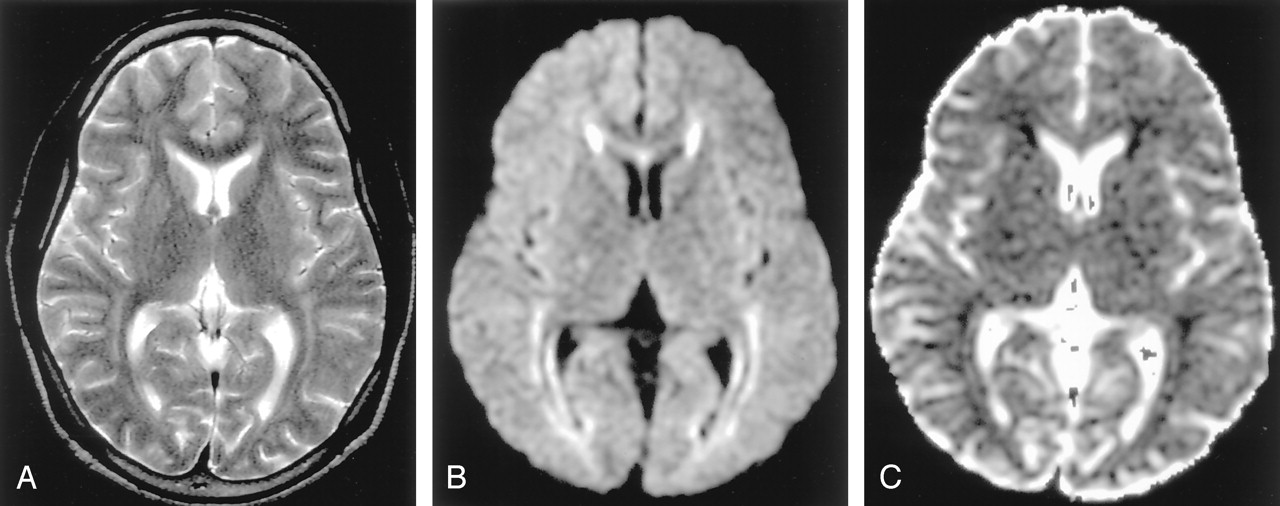
Phenylketonuria in a 36-year-old man. (From Moritani T, Ekholm S, Westesson PL. Diffusion-Weighted MR Imaging of the Brain. Berlin: Springer-Verlag; 2004. Used with permission.)
A, T2-weighted image shows hyperintense lesions in the periventricular white matter.
B and C, DW image shows these lesions as hyperintense with decreased ADC. Excitotoxicity may be related to impairment of glutamate receptor function by l-phenylalanine.
Some organic acid disorders are characterized by an accumulation of organic acids that share structural similarities with the excitotoxic amino acid glutamate (D-2.L-2,3 hydroxyglutarate, glutarate) (41). DW imaging can show diffuse hyperintensity with decreased ADC in the basal ganglia (Fig 17) or in cerebral white matter.

Glutaric aciduria in a 13-year-old male adolescent.
A, T2-weighted image shows hyperintense lesions in the bilateral globus pallidus (arrows) and diffusely in the white matter.
B and C, DW image shows these lesions as hyperintense with decreased ADC. Excitotoxic injury may be due to an accumulation of organic acids that share structural similarities with glutamate.
Wernicke Encephalopathy
Thiamine (vitamin B1) deficiency can cause Wernicke encephalopathy, characterized by confusion, ataxia, abnormal eye movements, and visual loss. Without thiamine, the Krebs and pentose phosphate cycles cannot metabolize glucose (42). The enzymatic inactivity leads to accumulation of intracellular glutamate. Cellular homeostasis soon fails, resulting in release of glutamate into the extracellular space (43). Pathologic findings include demyelination, edema, astrocytic swelling, and necrosis in the mammillary bodies, thalamic and hypothalamic nuclei, periaqueductal gray matter, walls of the third and floor of the fourth ventricle, and less commonly, the caudate nuclei, frontal, and parietal cortex. With treatment of intravenous thiamine, these lesions may dissipate.
DW imaging shows these lesions as hyperintense with decreased or increased ADC. Lesions with decreased ADC are thought to represent cytotoxic edema of neurons or astrocytes, whereas those with increased ADC can represent vasogenic edema (Fig 18) (44, 45). Either lesion can be reversible (46).

Wernicke encephalopathy in a 75-year-old man. (From Moritani T, Ekholm S, Westesson PL. Diffusion-Weighted MR Imaging of the Brain. Berlin: Springer-Verlag; 2004. Used with permission.)
A, FLAIR image shows a symmetrically hyperintense lesion in the hypothalami (arrow).
B and C, DW image shows isointense lesions with mildly increased ADC in the hypothalami that may represent vasogenic edema (arrow). Release of glutamate into the extracellular space may cause lesions in Wernicke encephalopathy.
Multiple Sclerosis
Glutamate excitotoxity damages not only neurons and astrocytes but also oligodendrocytes, myelin sheaths, and axons (47). Glutamate and aspartate are increased in the CSF of patients with acute multiple sclerosis (48). In an immunohistochemical study, active lesions of multiple sclerosis showed high glutamate production in macrophages and microglia near areas of axonal damage (49). Excitotoxicity in oligodendrocytes, myelin sheaths, and axons may result in cytotoxic plaques.
Cytotoxic plaques are rare and probably seen in the hyperacute or acute phase of multiple sclerosis. They are hyperintense on DW images, with decreased ADC (Fig 19). Pathologic findings of cytotoxic plaques show mainly intramyelinic edema.
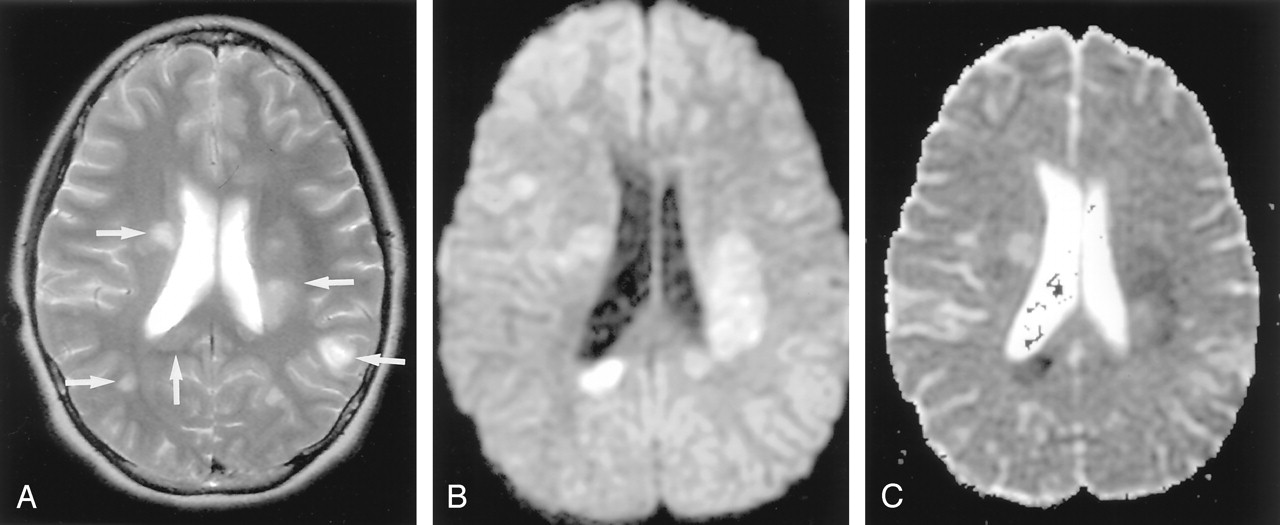
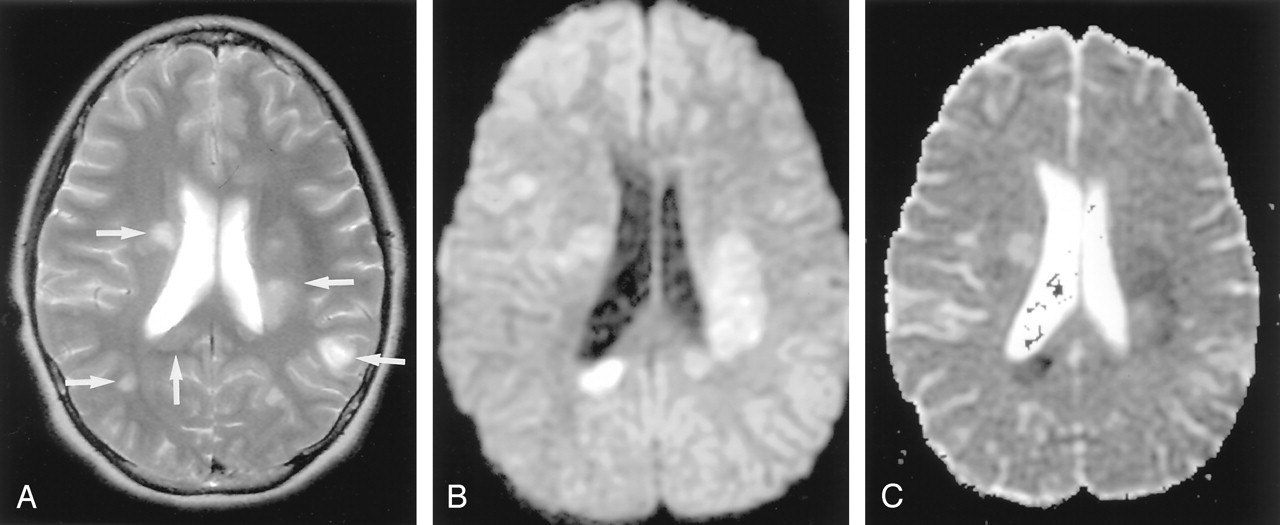
Acute multiple sclerosis in a 13-year-old female adolescent.
A, T2-weighted image shows multiple hyperintense lesions in the periventricular subcortical white matter and corpus callosum (arrows).
B and C, DW image shows some lesions as hyperintense with decreased ADC. They represent cytotoxic plaques presumably related to excitotoxic injury of oligodendrocytes, myelin sheaths, and axons.
Creutzfeldt-Jakob Disease
CJD is one of the prion diseases characterized by rapidly progressive dementia, myoclonus, and ataxia. Prion diseases are characterized by accumulation of misfolded prion protein, which is toxic to endoplasmic reticulum. Marked and selective abnormalities in glutamate receptors have recently been reported; these may explain the characteristic distribution of brain lesions in CJD (50).
T2-weighted images show hyperintense lesions in the cerebral cortex and bilateral basal ganglia in patients with CJD. The lesions often affect the bilateral thalami (pulvinar sign) and periaqueductal areas in patients with variant CJD (51, 52), but similar lesions can also be seen in sporadic CJD (53). DW imaging is more sensitive than conventional MR imaging for detecting abnormalities in CJD. DW imaging shows these lesions as hyperintense and often associated with decreased ADC (54–58) (Fig 20). Micropathologic findings show spongiform degeneration with 5–25-ìm vacuoles containing prion protein. Electron microscopy reveals that these vacuoles represent focal swelling of neuritic processes—both axonal and dendritic swelling (cellular edema)—that may cause decreased ADC (59).

CJD in a 51-year-old man with progressive dementia. (From Moritani T, Ekholm S, Westesson PL. Diffusion-Weighted MR Imaging of the Brain. Berlin: Springer-Verlag; 2004. Used with permission.)
A, T2-weighted image demonstrates mild hyperintensity bilaterally in the caudate nuclei, putamina, and pulvinar of the thalami (arrows).
B, DW image clearly demonstrates hyperintense lesions in these areas.
C, ADC is decreased in these lesions, which may represent cellular edema. The distribution of these lesions may be related to dysfunction of glutamate receptors.
Summary
DW imaging is useful for evaluating cytotoxic edema due to excitotoxic brain injury, a common final pathway for various neurologic diseases. The severity (reversibility) and distribution are different in various neurologic diseases (in terms of cell types, initial insults, and their mechanisms) and in patients of differing ages (with factors such as the age-dependent distribution of receptors and the maturity of the blood-brain barrier). Excitotoxic amine receptors are present in neurons, axons, glial cells, and myelin sheaths. Astrocytes and myelin sheaths, which protect synapses and axons, swell after absorbing excessive glutamate. Such cytotoxic edema seems to be transient and resolves on follow-up MR imaging.
Energy failure (impaired reuptake of glutamate) is the initial insult during infarction and HIE. It usually causes irreversible excitotoxic brain injury and typically results in necrosis and atrophy. Secondary degeneration, such as wallerian degeneration, seems to be related to excitotoxic circuits via the synapses or axons. Excessive release of glutamate can cause cytotoxic edema in conditions such as seizure, infection, demyelination, and toxic metabolic disease. Such cytotoxic edema is due to excitotoxic injury with less energy failure; this type occasionally resolves on follow-up MR imaging. Diffuse axonal injury can cause the leakage of glutamate from the axon at the node of Ranvier.
The distribution of lesions associated with status epilepticus seems to be related to the distribution of NMDA receptors. The severity and distribution in HIE, shaken baby syndrome, and neonatal herpes encephalitis seems to be related to increased vulnerability of the developing brain to excitotoxic injury.
In organic acid disorders, structurally similar substances can cause excitotoxic injury. Receptor dysfunction can occur in metabolic conditions (phenylketonuria) and degenerative diseases (CJD). Glutamate receptor antagonists may offer attractive possibilities for future therapy as possible neuroprotectants in the management of these diseases.
References
- Received July 14, 2004.
- Accepted after revision August 30, 2004.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Comprehensive Update and Review of Clinical and Imaging Features of SMART Syndrome
- Comprehensive Update and Review of Clinical and Imaging Features of SMART Syndrome
- Suspecting unwitnessed hypoglycaemia
- COVID-19 and Involvement of the Corpus Callosum: Potential Effect of the Cytokine Storm?
- Diffusion-Weighted MR Imaging in a Prospective Cohort of Children with Cerebral Malaria Offers Insights into Pathophysiology and Prognosis
- Cytotoxic edema affecting distinct fiber tracts in ciguatera fish poisoning
- Diffusion-Weighted Zonal Oblique Multislice-EPI Enhances the Detection of Small Lesions with Diffusion Restriction in the Brain Stem and Hippocampus: A Clinical Report of Selected Cases
- "Dazed and diffused": making sense of diffusion abnormalities in neurologic pathologies
- Anatomical patterns and correlated MRI findings of non-perinatal hypoxic-ischaemic encephalopathy
- Secondary Signal Change and an Apparent Diffusion Coefficient Decrease of the Substantia Nigra After Striatal Infarction
- Early Diffusion MR Imaging Findings and Short-Term Outcome in Comatose Patients with Hypoglycemia
- Diffusion MR Imaging of Hypoglycemic Encephalopathy
- Excitotoxicity in Acute Encephalopathy with Biphasic Seizures and Late Reduced Diffusion
- Shaken Baby Syndrome: Diagnosis and Treatment
- Focal splenial hyperintensity in epilepsy