
Abstract
BACKGROUND AND PURPOSE: The etiology of idiopathic normal-pressure hydrocephalus (NPH) is unknown. The purpose of this study was to examine the hypothesis that NPH begins in infancy as benign external hydrocephalus due to decreased uptake of CSF by the arachnoid villi. Since this occurs before the sutures fuse, a secondary hypothesis is that the intracranial volumes of patients with NPH should be larger than those of healthy individuals.
METHODS: Intracranial volumes of 51 patients with clinically suspected NPH were compared with those of age- and sex-matched control subjects. All patients underwent phase-contrast CSF velocity MR imaging. They had aqueductal CSF stroke volumes of at least 60 μL, which was 50% higher than previously published normal values. Intracranial volumes were measured and compared between groups.
RESULTS: The average intracranial volume for men with NPH (n = 22) was 1682 mL compared with 1565 for male control subjects (n = 55). The NPH volume averaged 118 mL (7.5%) larger than the control volume (P = .003). The average intracranial volume for women with NPH (n = 29) was 1493 mL compared with 1405 mL for female control subjects (n = 55). The NPH volume was 88 mL (6.3%) larger than the control volume (P = .002).
CONCLUSION: Patients with NPH have intracranial volumes significantly larger than normal, suggesting that the initial insult occurs before the sutures fuse at 1 year of age. The patients somehow remain asymptomatic until their later years, when a second insult must occur, leading to symptomatic NPH.
Normal-pressure hydrocephalus (NPH) was first described by Hakim (1) in 1964. NPH is characterized as communicating hydrocephalus in elderly patients presenting with the clinical triad of a gait disturbance, dementia, and incontinence. The original description focused on patients with idiopathic NPH (i.e., those without a known cause of communicating hydrocephalus) such as subarachnoid hemorrhage (SAH) or meningitis (2–4). Subsequently, chronic communicating hydrocephalus from these known causes has been added to the diagnosis of NPH. Patients with this form tend to be younger and have a better response to ventricular shunt surgery than those with the idiopathic form (5, 6).
A hypothesis was recently advanced to explain the cause of the idiopathic form of NPH. Specifically, patients with this disease were proposed to have had enlarged ventricles since infancy (7) owing to benign external hydrocephalus (8). This condition is thought to result from abnormal, presumably immature, arachnoid villi that cannot absorb enough CSF to keep up with the production. CSF then accumulates within the ventricles, because the sutures can still expand before 1 year of age over the convexities in an expanded, predominantly frontal, subarachnoid space (9). The arachnoid villi are thought to mature by 18 months of age, and head circumference no longer continues to increase but tracks parallel to the 95th percentile (10).
The hypothesis (7, 8) suggests that these patients remain asymptomatic with their slightly enlarged ventricles until their elderly years, when a second insult occurs, one possibly related to deep white matter ischemia (DWMI) and several associated conditions. Whether this second episode is due to decreased CSF resorption (7, 8), worsening periventricular ischemia (11, 12), softening of the brain (13), or another condition, the additional insult must lead to symptomatic NPH.
Ideally, to test this hypothesis, one would follow up children with benign external hydrocephalus to old age to see if they develop NPH. Clearly, this approach is impractical. However, if today’s NPH patients truly had benign external hydrocephalus as infants, their heads should be larger, reflecting expansion of the sutures in infancy. On the other hand, if NPH is a disease of only the elderly, their intracranial volumes should not be larger than those of control subjects. The purpose of this study was to compare intracranial volumes, as shown by MR imaging, in patients with clinical NPH with those of age- and sex-matched control subjects. We hypothesized that the patients with NPH have larger intracranial volumes.
Methods
A total of 66 consecutive patients with clinically suspected idiopathic NPH were evaluated by means of MR imaging over the 2-year period from 2000 to 2002. All patients had been referred by neurologists or neurosurgeons and had symptoms of gait disturbance and dementia. All patients gave informed consent for their studies to be used (anonymously) for research. The review board of the institution where the images were acquired approved the use of these data.
Conventional MR imaging of the brain was performed, including 5-mm-thick T1-weighted sagittal spin-echo (SE) images (TR/TE, 500/15) and proton density–weighted and T2-weighted axial SE images (3000/20 and 80). Both sequences had a 1-mm intersection gap and were conventional SE acquisitions rather than fast SE. A 256 × 192 matrix was acquired over a 24 × 16-cm field of view. Because this was a retrospective study, a 3D acquisition without a gap was not acquired.
Phase-contrast CSF velocity imaging was performed to measure the aqueductal CSF stroke volume (i.e., the volume of CSF pulsating back and forth through the aqueduct) over the cardiac cycle (14). A 4-mm-thick section was angled perpendicular to the aqueduct by using a 512 × 512 matrix over a 16 × 16 cm field of view, yielding pixels of 312 μm on a side. Retrospective cardiac gating and an aliasing velocity of 200 mm/s were used (14). Only patients with hyperdynamic CSF flow were included in the study group, leaving 51 patients. Hyperdynamic CSF flow was defined as a stroke volume greater than 60 μL, which is approximately 50% higher than the normal value reported in the literature (14). In patients with suspected NPH in whom CSF velocity imaging was technically flawed because of cardiac arrhythmia or velocity aliasing, hyperdynamic CSF flow was diagnosed on the basis of a marked aqueductal CSF flow void on the proton density–weighted conventional SE image (15). As noted previously (14), the CSF flow void is less sensitive than originally described (15) because of the ubiquitous use of flow compensation for the last 2 decades and fast SE for the last 10 years. Regardless, when the flow void is seen, it is specific for hyperdynamic CSF flow. Therefore, the NPH group comprised 22 men and 29 women with clinically suspected NPH and hyperdynamic CSF flow.
For the control group, age- and sex-matched control subjects were chosen from 110 elderly patients who underwent imaging in 2001 and 2002. These patients were consecutive unless they had clearly enlarged ventricles (possible preclinical NPH) or evidence of previous stroke or other encephaloclastic process. The average age of the men with NPH was 75 years (range, 62–86 years) compared with 71 years (range, 59–88 years) for male control subjects. The average age of the women with NPH was 77 years (range, 61–88 years) compared with 74 years (range, 57–96 years) for the female control subjects. The average stroke volume was 149 μL for men with NPH and 127 μL for women with NPH. The normal value obtained by using an identical technique is 42 μL, as reported in the literature (14).
T2-weighted axial images from the NPH and control groups were transferred in Digital Imaging and Communications in Medicine, or DICOM, format to a workstation. The interpolated, smoothed boundaries of the hyperintense CSF were traced at the widest portion of the brain on the original axial images, as well as on reformatted sagittal and coronal images. By using tools on the workstation (maximum intensity projection, surface, and measure), the three planar sections were rendered into a 3D surface reconstruction. The intracranial volume was measured by using an automated 3D technique, which consisted of counting voxels within the defined surface and multiplying it by voxel volume (Fig 1). The NPH and control groups were mixed together to avoid bias in the volume measurement. Intraobserver variability was measured in 17 patients.

Measurement of intracranial volume. Hyperintense CSF is outlined on T2-weighted images, and volume is calculated. Gaps have been interpolated to provide smooth surface contours.
A, Axial image.
B, Reformatted sagittal image.
C, Reformatted coronal image.
A phantom consisting of 1500 mL of normal sodium chloride solution was imaged before and after the removal of 75 mL by using the same T2-weighted SE technique (with 5-mm section thickness and a 1-mm gap) as that used for patients and control subjects. The volume was confirmed by using a graduated cylinder. Volume measurements were performed on three separate occasions to determine the accuracy of this technique.
Results
The absolute mean error in the measurement of the 1500- and 1425-mL phantoms was 5.0 mL (range, 0.2–10 mL), which represented 0.3% error. The average of the three measurements for the 1500-mL phantom was 1495 mL, and the average for the 1425-mL phantom was 1426.4 mL. Therefore, the following volumetric measurements were considered sufficiently reproducible, precise, and accurate for the current study.
The average intracranial volume for the 22 men with NPH was 1682 mL (SD = 148) compared with 1565 mL (SD = 164) for the 55 male control subjects. The average NPH volume for the men was 118 mL (7.5%) larger than the control volume; the difference was statistically significant (P = .003). The average intracranial volume for the 29 women with NPH was 1493 mL (SD = 123 mL) compared with 1405 mL (SD = 107 mL) for the 55 female control subjects. The average NPH volume for the women was 88 mL (6.3%) larger than the control volume; this difference was statistically significant (P = .002). The average intraobserver variability was 12 mL (0.7%) for men (n = 9) and 22 mL (1.5%) for women (n = 8).
Discussion
If NPH truly does begin as benign external hydrocephalus in infancy, it would essentially be a completely treatable disease—if our healthcare system could afford to image every patient as they approach 60 years of age. Because this is obviously impractical and cost-prohibitive, we might at least follow up patients with previously diagnosed slightly enlarged ventricles for symptoms of NPH, as early shunt surgery may prevent the progression of symptoms.
The original description of NPH considered the disease to be idiopathic (i.e., with no evidence of previous SAH or meningitis and no history of trauma) (3, 4). NPH is diagnosed when elderly patients presenting with the clinical triad of gait disturbance, dementia, and incontinence have radiographic evidence of communicating hydrocephalus (i.e., ventricular enlargement out of proportion to cortical sulcal enlargement) (1, 2). This ventricular enlargement has always been presumed to occur later in life, at the time of clinical presentation. The mechanism of enlargement has been attributed to DWMI, which a number of authors have shown to be increased in patients with NPH (16, 17).
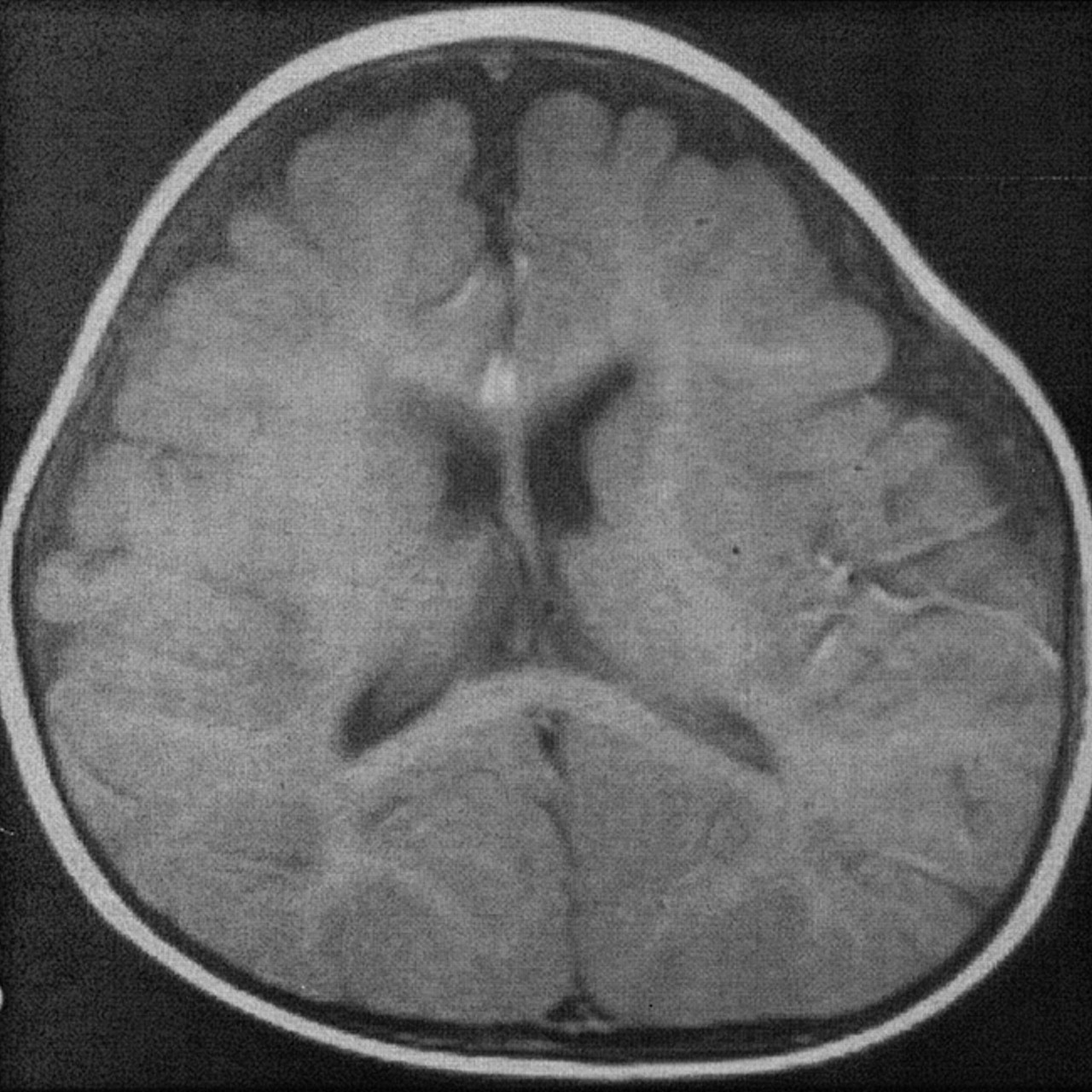
Several years ago, the theory was advanced that patients with NPH might not develop ventricular enlargement as elderly adults, but rather, they have had enlarged ventricles since infancy (7), when they had benign external hydrocephalus (8). Modern pediatricians detect this condition on the basis of increasing head circumference that is out of proportion to increases in body weight and length (9, 10). The cause is supposedly immature arachnoid villi that cannot resorb CSF as quickly as it is produced by the choroid plexus. As a result, the CSF backs up. Because the sutures are still open in these infants who are less than 1 year old, CSF accumulates both within the slightly enlarged ventricles and over the convexities in an enlarged, predominantly frontal, subarachnoid space (Fig 2). Some have stressed that radiologists must recognize this entity, as it is “fairly common” (18) and self-limited, and children thus affected do not require ventricular shunt surgery. As the name suggests, the condition is supposedly benign (9, 10).

Benign external hydrocephalus in 7-month-old infant results in enlargement of the frontal subarachnoid spaces with minimal ventricular enlargement.

Chronically enlarged ventricles in a patient developing NPH.
A, At age 67 years, this patient had clearly enlarged ventricles. CT was performed to evaluate suspected giant cell arteritis. The patient was walking 20 miles a day and underwent successful shunt placement for NPH 19 years later.
B, At age 70, the patient still had no symptoms of NPH.
C–E, At age 76, the ventricles are markedly enlarged. Proton density–weighted image (C) shows a prominent CSF flow void in the aqueduct. The patient will not develop symptoms of NPH for another 10 years (when a pacemaker precluded MR imaging).
Results of the current study suggest that benign external hydrocephalus may be less benign than previously thought. The statistically larger intracranial volumes of NPH patients compared with age- and sex-matched control subjects suggest that the process begins in infancy. Because pediatricians were not routinely checking head circumferences 60–80 years ago, there was never any indication that there was an abnormality. MR imaging and CT studies of the brain performed for reasons other than NPH evaluation in adulthood may have shown slightly enlarged ventricles; these were interpreted as being at the upper limits of normal, as the usual causes of communicating hydrocephalus (SAH and meningitis) were not part of the clinical history. It now appears that patients with such findings may be at risk for developing NPH in their later years.
Benign external hydrocephalus is characterized by decreased CSF resorption. Because MR imaging shows no evidence of a brain tumor or other cause of obstruction, the evaluation is usually concluded with this benign diagnosis. Given the apparent benignancy, none of the children ever undergo a saline infusion test (requiring lumbar puncture), which would continue to demonstrate decreased CSF resorption. Only 60–80 years later, when they are being evaluated for a gait disturbance, dementia, and possible NPH, do they undergo a saline infusion test, which shows decreased CSF resorption (19, 20). The fact that histologic analysis of the meninges in NPH fails to show evidence of SAH or meningitis (21) is supportive of this theory. These findings also raise the question: Are the arachnoid villi truly immature in benign external hydrocephalus or will these patients always have decreased CSF resorptive capacity? Lack of increasing head circumference after 18 months could be indicative of sutural closure rather than maturation of the arachnoid villi.
The specific event that leads to decompensation in later life is not entirely clear. It is now well documented that there is a statistically higher incidence of DWMI in patients with NPH compared with age-matched control subjects; therefore, it is presumably somehow related to that process (16, 17, 22–27). At least three potential mechanisms mediated by or related to DWMI could lead to the clinical decompensation in NPH: increasing periventricular ischemia, softening of the brain, and decreased CSF resorption. It is also possible that a combination of all three mechanisms contributes to the final insult.
Given the larger ventricles of patients with NPH compared with those of infants with benign external hydrocephalus, it is likely that the ventricles have been slowly enlarging throughout life without symptoms (Fig 3). Only in later life is a functional threshold surpassed, leading to clinical decompensation. Each of these three potential mechanisms is discussed next.
DWMI is known to be associated with increased ischemia of the periventricular white matter (11). Normally, intravenous administration of the carbonic anhydrase inhibitor acetazolamide (e.g., Diamox, Lederle Labs, Pearl River, NY) causes increased cerebral blood flow due to arteriolar dilatation. In patients with NPH, there is no increase in cerebral blood flow, particularly in the deep white matter, suggesting that the arterioles are already maximally dilated. Perhaps the symptoms in NPH occur not because of additional ventricular enlargement but because of the insult of worsening ischemia. Although this explanation might lead to symptoms, it does not lead to progressive ventricular enlargement.
A second possibility for the second insult in NPH is changing viscoelastic properties (i.e., softening or increased compressibility of the brain) due to DWMI (13, 28). While it has always been presumed that the ventricles pulsate outwards against the paracentral white matter tracts, in fact, phase-contrast MR studies show that systolic expansion of the brain causes it to compress the ventricles during systole (29). The greatest shearing is at the boundary of the brain and the ventricles; therefore, it is not unexpected that progressive softening of the brain leads to fissuring in the immediate periventricular region, leading subsequently to additional ventricular enlargement. Thus, a vicious cycle ensues with progressive ventricular enlargement leading to increased interstitial pressure, increased capillary closure, increased ischemia, and increased softening.
Consideration of possibly decreased CSF resorption as a third cause of progressive ventricular enlargement necessitates a discussion of normal water movement in the brain. Water normally leaves the arterioles and enters the extracellular space of the brain under pressure and osmotic gradients (8). It may then either re-enter the venous side under an osmotic gradient or enter the subarachnoid space under a pressure gradient. (The osmotic effect is the reason that hyperosmolar mannitol decreases brain edema acutely.) Pressure gradients cannot be invoked as the cause of venous-side water re-entry since they would cause collapse of the capillaries and veins.
Excess water in the form of vasogenic edema from blood brain–barrier breakdown flows centrally through the extracellular space to be absorbed by the ventricles. Excess water crossing the ependyma as interstitial edema from obstructed ventricles flows peripherally through the extracellular space, eventually emptying into the subarachnoid space. Although this extracellular passage of water has occasionally been incorrectly referred to as the parenchymal CSF resorption route (8, 30–34), it in fact represents flow through the extracellular space of the brain rather than resorption by the parenchyma per se. This is probably the primary CSF resorption pathway in patients with long-standing obstruction (e.g., from tectal gliomas) who have complete obstruction to CSF flow through the aqueduct (33).
Patients with decreased CSF resorption via the arachnoid villi are more heavily reliant on resorption via the extracellular space of the brain. If the current theory is correct, infants with benign external hydrocephalus have been in a delicate equilibrium their entire lives, the amount of CSF produced being balanced by the amount of CSF being resorbed. Any increased resistance to CSF flow through the extracellular space could potentially upset this equilibrium.
The bulk motion of free water through the extracellular space of the brain is determined by the pressure gradient and the resistance to flow. With DWMI there is loss of myelin, axons, and oligodendroglial cells (35). The loss of lipid causes the environment to become more hydrophilic. As the lipid content of the deep white matter decreases, the water content increases. This is the reason that DWMI is hyperintense on T2-weighted images and that the apparent diffusion coefficient is increased on diffusion-weighted images (36).
With increasing hydrophilia of the deep white matter, there is increased hydrogen bonding of polar water molecules to the charged side groups of the myelin protein. However minimal it might be, this is certain to increase the resistance to movement of free water through the extracellular space of the brain, similar to the slowed progression of a chemical species with greater adsorption to the solid phase in a chromatographic column (37). With increasing resistance, there will be a tendency for water or CSF to back up in the ventricles, upsetting the delicate balance, leading to subtle worsening in the hydrocephalus.
If DWMI-mediated increased resistance to extracellular CSF flow is the dominant mechanism of ventricular enlargement beyond that due to benign external hydrocephalus, it would imply that the ventricles are only slightly enlarged until approximately age 60 years when DWMI begins. (This is consistent with the observation that only slightly enlarged ventricles are commonly observed as an incidental finding in children and adults, rather than the greater ventricular enlargement seen in NPH.) After approximately age 60 years, progressive ventricular enlargement should occur until the combination of increasing barotrauma, brain softening, and periventricular ischemia finally results in symptoms.
In the mathematical model of Rekate et al (13), increasing brain compressibility or softening, plus increased resistance to CSF outflow to the subarachnoid space was shown to lead to ventricular enlargement. Given that all other pathways of CSF egress from the ventricles to the SAS are presumably fixed in patients with NPH, increasing resistance to flow through the extracellular space should also contribute to ventricular enlargement.
Regardless of the specific mechanism of the second insult, it seems clear that patients with enlarged ventricles are at risk for NPH in their later years as DWMI worsens. That being said, patients with known ventricular enlargement of any cause should be watched carefully for early signs of gait disturbance, indicating onset of NPH. (Perhaps such patients would benefit from functional MR imaging performed during visualization of walking, since patients with NPH presumably require more mental energy and brain activation to accomplish the task than healthy subjects [38].) Following the onset of symptoms, shunt surgery should be considered in patients with NPH early in the course of the disease before they have progressed to the point where atrophy is present.
There were several potential problems with this study. The diagnosis of NPH was not confirmed by response to shunt surgery. Although hyperdynamic CSF flow has been shown to be correlated with the response to shunt surgery in patients with symptomatic NPH (14, 15), the surrogate measure is less convincing than shunt response per se (which was unfortunately not available). The height and weight of the patients were also not available to adjust for head size; the only mechanism to account for different body sizes was to compare men and women separately.
Regardless of the shortfalls of this study, it appears that NPH might not be considered a disease of only the elderly. NPH is estimated to account for 1–10% of all patients with dementia, and it may affect as many as 350,000 elderly adults (39). Given that the ventricles may have been enlarged all along, NPH should be a largely preventable disease.
Conclusion
The results of this study suggest that the etiology of idiopathic NPH may be benign external hydrocephalus in infancy. The finding that intracranial volume is significantly larger in age- and sex-matched control subjects supports this hypothesis. If borne out by subsequent longitudinal studies, these findings would suggest that NPH is a largely treatable disease.
Acknowledgments
We thank neurosurgeons Gordan McComb, MD, and Harold Rekate, MD, for their helpful discussions and David Dang, MD, for his help with the statistical analysis.
References
- Received January 2, 2004.
- Accepted after revision March 17, 2004.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Molecular Signatures of Normal Pressure Hydrocephalus: A Large-scale Proteomic Analysis of Cerebrospinal Fluid
- Aqueductal CSF Stroke Volume Is Increased in Patients with Idiopathic Normal Pressure Hydrocephalus and Decreases after Shunt Surgery
- Lumbar Puncture Test in Normal Pressure Hydrocephalus: Does the Volume of CSF Removed Affect the Response to Tap?
- CSF Flow in the Brain in the Context of Normal Pressure Hydrocephalus
- Evidence that congenital hydrocephalus is a precursor to idiopathic normal pressure hydrocephalus in only a subset of patients
- Idiopathic normal pressure hydrocephalus