
Abstract
BACKGROUND AND PURPOSE: The middle interhemispheric variant of holoprosencephaly (MIH) is a rare malformation in which the cerebral hemispheres fail to divide in the posterior frontal and parietal regions. We herein describe the structural abnormalities of the brain in a large group of patients with MIH, compare these features with those of classic holoprosencephaly (HPE), and propose a developmental mechanism, based on current knowledge of developmental neurogenetics, by which MIH develops.
METHODS: Brain images obtained in 21 patients with MIH (MR images in 16 patients and high-quality X-ray CT scans in five patients) were retrospectively reviewed to classify cerebral abnormalities. The cerebral parenchyma, hypothalami, caudate nuclei, lentiform nuclei, thalami, and mesencephalon were examined for the degree of midline separation (cleavage) of the two hemispheres. The orbits, olfactory apparati, and presence or absence of a dorsal cyst were also assessed.
RESULTS: In all patients, by definition, midportions of the cerebral hemispheres were continuous across the midline, with an intervening interhemispheric fissure. The sylvian fissures were abnormally connected across the midline over the vertex in 18 (86%) of 21 patients. Two patients had relatively normal-appearing sylvian fissures; one had unilateral absence of a sylvian fissure owing to substantial subcortical heterotopia. Heterotopic gray matter or dysplastic cerebral cortex was also seen in 18 (86%) of 21 patients. MIH differed from classic HPE as follows. 1) In all subjects, the midline third ventricle separated the hypothalamus and lentiform nuclei. 2) The caudate nuclei were separated by the cerebral ventricles in 17 (89%) of the 17 patients in whom they could be assessed. 3) The most commonly affected basal nucleus was the thalamus (non-cleavage in seven [33%] of 21 cases, abnormal alignment in 1 [5%]). 4) Three (18%) of the 16 patients in whom the mesencephalon could be assessed showed some degree of mesencephalic non-cleavage. 5) No patients had hypotelorism (four had hypertelorism, the remainder manifested normal intraocular distances). Dorsal cysts were present in five (25%) of the 20 patients in whom they could be assessed (dorsal cysts could not be assessed after shunt surgery), and as in classic HPE, were associated with severe thalamic non-cleavage in three of these five patients.
CONCLUSION: MIH appears to cause non-cleavage of midline structures in a completely different pattern than does classic HPE. In MIH, impaired induction or expression of genetic factors appears to influence the embryonic roof plate, whereas in classic HPE, induction or expression of the embryonic floor plate seems to be affected.
The middle interhemispheric variant of holoprosencephaly (MIH) was first described in 1993 (1). Also referred to as “syntelencephaly,” this malformation consists of an abnormal midline connection of the cerebral hemispheres in the posterior frontal and parietal regions, with interhemispheric separation of the basal forebrain, anterior frontal lobes, and occipital regions. Since the first report, additional case reports and small series describing this unusual malformation have appeared in the literature (2–5). To our knowledge, however, no investigation assessing structural abnormalities in MIH in a large patient series has been published. A detailed analysis of MIH and a comparison of its features with those of classic forms of holoprosencephaly (HPE) (recently reported in detail in two large series [6, 7]) could be valuable in clarifying the underlying developmental processes in both. Briefly, three classic forms of HPE have been described. Alobar HPE, the most severe of the three types, generally appears as a pancake-like mass of cerebral tissue and a crescentic monoventricle. Semilobar HPE is characterized by non-cleaved anterior brain, with the posterior falx and posterior interhemispheric fissure (IHF) present. Lobar HPE is also characterized by anterior brain non-cleavage that exists to a lesser degree than that of semilobar HPE, a fully formed third ventricle, and some development of frontal horns of the lateral ventricles (8–10).
Herein we provide a detailed analysis of brain images obtained in 21 patients with MIH, compare these findings with those observed in classic HPE, and correlate these data with the most current information regarding the genetic factors controlling early brain development. Our focus centers on those regions of the developing brain involved in dorsal-ventral and medial-lateral patterning.
Methods
Brain images were retrospectively selected from a group of patients referred to The Carter Centers for Brain Research in Holoprosencephaly and Related Malformations as well as from those patients referred to us at our respective institutions. The Carter Centers is a national consortium funded by a not-for-profit foundation, with its primary clinical research and treatment center at the Texas Scottish Rite Hospital for Children (Dallas, TX). A total of 121 brain images were retrospectively reviewed by two neuroradiologists (E.M.S., A.J.B.). One hundred of these patients manifested classic HPE according to standard definitions (8–10), and a subset of these has been reported on previously (6, 7). MIH was diagnosed in the remaining 21 patients (age range, 1 day to 4 years old). Two of these cases have been described in an earlier publication (1).
All 21 patients had adequate imaging studies for assessment of most of the components of the malformation spectrum. The images that were used included MR images obtained in 16 patients and high-quality X-ray CT scans obtained in five patients. Because images were obtained at multiple institutions on a variety of imaging systems, techniques varied considerably. All MR imaging included sagittal T1-weighted sequences and at least one examination in the axial plane with either a T1- or T2-weighted sequence. Coronal sections were available in 14 patients. Paramagnetic contrast agent was not routinely administered. CT scans were only obtained in the axial plane and no contrast agent was used.
The caudate nuclei, lentiform nuclei, thalami, and thalamic orientation were graded according to a previously presented scale (6). The sylvian fissures were analyzed with respect to location and orientation. The portions of the corpus callosum that were present were documented, as determined by assuring that the commissure connected neocortical white matter of each hemisphere. Three of the five CT scans allowed adequate assessment of the callosal genu and spenium. The callosal body, which parallels the image plane on axial images, cannot be accurately assessed by CT alone.
The intraocular distance (IOD) was determined by correlating the interpupillary distance (IPD) and bony interorbital distance (BIOD) with patient age (11). The presence or absence of the olfactory sulci and bulbs was noted; if present, subjective determination of normal versus reduced size was made. The structure of the cerebellum was subjectively assessed.
When visible, the arteries of the anterior cerebral circulation were evaluated for alterations in pattern, particularly the presence of an azygous anterior cerebral artery. Also, when imaging allowed, the pituitary gland was subjectively graded as normal or abnormal in size and signal intensity for age (12).
Results
The sylvian fissures were abnormally connected across the midline over the vertex in 18 (86%) of 21 patients. Two patients had relatively normal-appearing sylvian fissures; one had no right sylvian fissure owing to a large subcortical heterotopia. Heterotopic gray matter or cortical dysplasia were also seen in 18 of 21 cases that were not necessarily associated with abnormal sylvian fissures, including abnormal thickening of the cortex lining the anterior IHF. Reduced sulcation (ie, the normal cingulate sulci and internal frontal sulci were absent) in 15 (71%) of the 21 patients (Figs 1 and 2).

Axial T1-weighted image (550/14/2 [TR/TE/excitations]) in the posterior frontal and parietal regions shows the abnormal sylvian fissures communicating across the midline over the vertex (black arrows). Flow-related enhancement is seen within branches of the middle cerebral arteries, confirming that this represents the sylvian fissure. Thickened dysplastic cortex is present along the anterior IHF (white arrows).

Coronal echo-planar inversion recovery image (2000/30/400 [TR/TE/TI]) depicts the large area of subcortical dysplasia and associated gray matter heterotopia (white arrows). The site of the repaired cephalocele can be seen at the vertex (black arrows). The inferior vermis is absent.
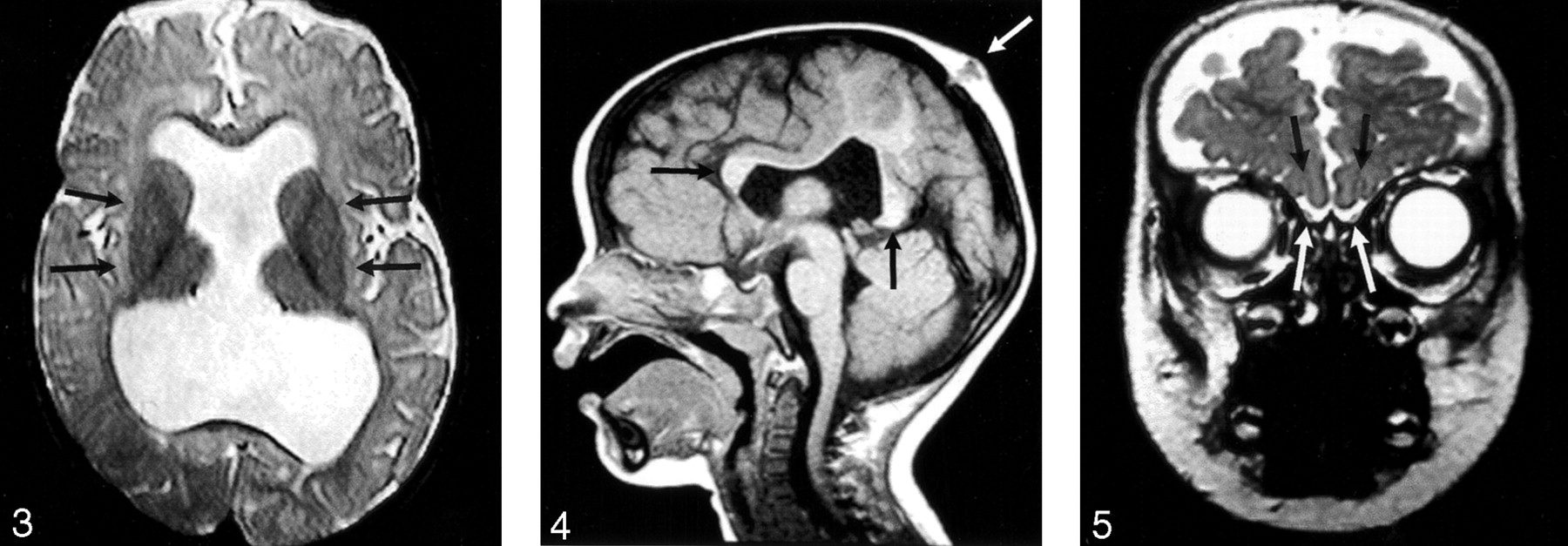
The hypothalamus and lentiform nuclei appeared normally separated in all patients. The caudate nuclei were incompletely separated in two (11%) of the 19 patients in whom they could be adequately assessed, whereas the thalami could be visualized in all 21 patients and were incompletely separated in seven (33%) (Fig 3). The thalami were abnormally oriented, with their long axes nearly parallel, in only one (5%) patient. The mesencephalon appeared incompletely separated from the diencephalon in three (18%) of the 17 patients in whom this could be assessed. Dorsal midline cysts were present in five (25%) of 20 MIH patients in whom it could be assessed (could not be assessed if images were acquired after shunt surgery) and was associated with severe thalamic non-cleavage in three of these five patients.

Axial spin-echo T2-weighted image (2530/80/2 [TR/TE/excitations]) shows the widely separated deep gray nuclei (arrows), unlike those seen in classic HPE.
Some component of the corpus callosum was present in all 18 patients in whom the corpus could be reasonably assessed (assessment was not possible in three patients who had only undergone CT scanning). The splenium and genu were identified in 11 (61%) of 18 (Fig 4). Four (22%) of these 18 patients had either a splenium or genu (but not both structures), and three (19%) of the 16 patients who underwent MR imaging had some portion of the body present in addition to the splenium of the callosum or genu of the callosum or both. No patient had a complete corpus callosum.

Sagittal T1-weighted image (400/15/1) through the midline reveals the genu and splenium of the corpus callosum (black arrows). The body of the callosum is absent in the region of non-cleaved parenchyma. An atretic cephalocele is also present (white arrow). Note also that the anterior recess of the third ventricle and basal forebrain are normal, unlike that seen in classic HPE.
Analysis of the orbits revealed a normal IOD in 13 (77%) of the 17 patients in whom it could be accurately examined. Four patients had hypertelorism (IPD and BIOD at or above the 97th percentile for age). It was also noted that, in those with a “normal” IOD, all patients were at or above the 50th percentile for age. No patients had hypotelorism. In 12 (92%) of 13 of the cases in which it could be assessed, the optic chiasm was normal in appearance. One patient had a small (presumably hypoplastic) chiasm.
The anterior cerebral arterial circulation pattern could be adequately assessed in 16 of our 21 patients. In all 16 patients, the anterior cerebral artery was unpaired (azygous), whereas the middle cerebral artery trunks appeared normal.
The inferior frontal lobes could be assessed in the coronal plane in 14 cases, allowing for the evaluation of the olfactory sulci in all 14 and the olfactory bulbs in 11. Olfactory sulci were present and normal in configuration in eight (57%) of 14 cases. The remainder were absent (2) or present but shallow (presumably hypoplastic) (4). Olfactory bulbs appeared normal in size and location in seven (64%) of 11 cases (all seven with normal olfactory sulci) (Fig 5). The remaining four appeared small.

Coronal fast spin-echo T2-weighted image (3200/117/1) through the inferior frontal lobes depicts the normal appearance of the anterior IHF, olfactory sulci (black arrows), and olfactory bulbs (white arrows).
Two (10%) of the 21 patients were noted to have cephaloceles overlying the “bridge” of unseparated parenchyma. One was atretic, whereas the other was large and required surgical intervention (Figs 2 and 4).
Abnormalities of the cerebellum were present in four (19%) of 21 patients, including Chiari I and II malformations, cerebellar hypoplasia, and inferior vermian hypogenesis (Fig 2).
Sagittal and coronal MR imaging allowed examination of the pituitary gland in 16 patients. The gland was seen in all patients; it was normal in 12 and subjectively small in four.
Discussion
This analysis of 21 patients suggests that the components of the MIH malformation differ substantially from those seen in classic HPE. The relative sparing of the basal forebrain with a well-formed anterior IHF, the greater involvement of the thalami than that in the caudates and hypothalami, the different locations of callosal involvement, and the overwhelmingly normal IOD and optic chiasm formation all suggest that MIH either has a different embryogenetic mechanism than that of classic HPE or has a similar mechanism that acts at a different location.
However, both MIH and classic HPE share a fundamental similarity: non-cleavage of a substantial portion of the supratentorial brain into two separate hemispheres. In addition, several other features are seen in both MIH and classic HPE, including the abnormalities of the anterior arterial circulation and sylvian fissure structure. Thus, it seems that some underlying similarity must exist in the mechanisms by which these malformations develop.
Some insight into the embryologic development of MIH was provided by the recent discovery that one of the patients included in this study has a mutation of the ZIC2 gene. The human ZIC2 gene encodes a zinc finger transcription factor, a DNA-binding, gene-specific factor that consists of 28 amino acid repeats, each sequence folded around a zinc ion (13). Studies of the homologous mouse gene, Zic2, have indicated that Zic2 plays important roles in neural tube closure and in differentiation of the roof plate of the developing embryo (Fig 6). In mice, mutation of Zic2 results in defects of neural tube closure and HPE. Both of these defects seem to result from impaired expression of roof plate-specific properties, such as decreased mitotic rate and increased apoptotic rate (14). The reduction of mitotic rate and increase of apoptosis in the roof plate are critical to the formation of the IHF. Thus, mutation of Zic2 may result in a continuum of cerebral tissue across the midline of the dorsal cerebrum, without an intervening IHF (Fig 6), precisely the structural appearance of MIH.

Illustration of roof plate differentiation, which depends on ZIC2 function.
After neural tube closure (left), ZIC2 promotes differentiation of the embryonic roof plate (rp) in the dorsal midline of the neural tube. Normally, a low rate of mitotic activity and a high rate of apoptosis in the roof plate of the forebrain contribute to interhemispheric cleavage (upper right). If one ZIC2 allele is mutant (ZIC2+/-), differentiation of the roof plate is compromised, resulting in HPE (lower right).
cx indicates cortex; ey, eye; fp, floor plate; hy, hypothalamus; st, striatum.
In contrast to other genes that have been linked to HPE (eg, Sonic hedgehog), ZIC2 is not involved in patterning of the embryonic dorsoventral axis but instead acts in the dorsal midline to promote differentiation of the roof plate. This difference may explain the absence of craniofacial malformations and the occurrence of middle interhemispheric variants in HPE cases linked to ZIC2 mutations. Because dorsal structures are primarily affected, effects on ventral structures, such as hypotelorism and lack of formation of the medial aspects of the hypothalamus and caudates (morphologic characterisics of more classic forms of HPE [6]) are less likely. Indeed, presumably because Zic2 is involved in the differentiation of the roof plate and not of the floor plate, the craniofacial structures are not malformed in mice with Zic2-related HPE (14), nor in humans with ZIC2-related HPE (13). Our findings of above average IODs or hypertelorism in all 21 patients further support this difference from classic HPE. Also, because effects of ZIC2 deficiency may occur segmentally along the rostrocaudal axis of the neural tube, cases with MIH may represent instances in which the midportion of the forebrain was selectively vulnerable to the genetic defect. In future studies, it will be important to verify these conclusions by correlating the phenotypic effects in HPE with genetic and other bases.
It is intriguing to note that Zic2 is associated with neural tube closure, the failure of which may result in cephalocele, myelomeningocele, or Chiari II malformation, and that cephaloceles and Chiari II malformations were both present in some of our cases. We believe the presence of these associated malformations further strengthens the likely association between ZIC2 and MIH. Indeed, it was our impression that patients with MIH had a higher incidence of substantial cerebellar anomalies (such as Chiari malformations) than those with classic HPE. In our experience, approximately 10% of patients with classic HPE have an associated cerebellar abnormality and most (>80%) are mild inferior vermian hypogenesis (Simon EM, unpublished data).
The abnormal sylvian fissures, with their abnormal angulation and communication across the midline over the vertex seems to have been previously reported. One report labeled this finding as a “connected transhermispheric cleft of focal cortical dysplasia” in a patient with HPE (2). A review of the images published in their case report reveals multiple vascular branches in the sulcus. We believe that these are branches of the middle cerebral arteries, which would clearly show these as extensions of the sylvian fissures. Another report (5) of a nearly identical case described two siblings with “fused schizencephalic clefts in their bilateral frontal lobes.” A similar finding was present in the images accompanying the case reported by Robin et al (4), but the authors did not specifically mention this component of the malformation. By reviewing images from a larger series of MIH patients, we were able to identify this pattern consistently and realize that the sulci are extensions of the sylvian fissures and are common structural characteristics of MIH.
Our review of the literature also revealed case reports that were described as “lobar” HPE, but review of the published figures or pathologic descriptions suggests that these were cases of MIH (15, 16). An intact anterior IHF can be identified in the images presented, and the deep gray nuclear and ventricular configurations closely resemble those seen in our series (15). These two cases have been reported as a constellation of HPE, hypertelorism, and ectrodactyly. To our knowledge, our patients with MIH and hypertelorism did not have anomalies of their hands.
The advances of modern medicine allow patients with severe brain malformations to survive infancy, often into late childhood and adulthood. Therefore, it is useful to be able to diagnose brain malformations and prognosticate developmental potential of affected infants. Accurate diagnosis allows proper counseling of parents and preparation of caregivers and parents for the future needs of the child. Correlation of specific morphologic features of these anomalies with outcome may allow identification of specific features that make such prognostication possible. Thus, such detailed analysis of structural features of dysmorphic brains may allow prognostication and elucidate potential embryogenetic mechanisms. Future investigations are planned to correlate these morphologic abnormalities with specific clinical features and generate more accurate prognostic information.
Conclusion
Our analysis of these MIH patients further refines the morphologic spectrum of this disorder and further contrasts it with the structural characteristics of classic HPE. These morphologic differences allow further elucidation of the embryologic differences between the two malformations, one likely resulting from the disturbances of formation of the embryonic floor plate (classic HPE) and the other of the roof plate (MIH). Future correlation of the morphologic data with clinical and neurodevelopmental information may help to provide more accurate prognostic information to families of patients with MIH.
Footnotes
Funded in part by the Carter Centers for Brain Research in Holoprosencephaly and Related Malformations; J.D.P. supported by NIH Grant No. K12 NSO1692.
Presented at the 39th Annual Meeting of the American Society of Neuroradiology, Boston, MA, April 2001.
References
- Received September 18, 2001.
- Accepted after revision August 1, 2001.
- American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Monozygotic twins with de novo ZIC2 gene mutations discordant for the type of holoprosencephaly
- Asymptomatic Interhypothalamic Adhesions in Children
- In Utero MR Imaging of Fetal Holoprosencephaly: A Structured Approach to Diagnosis and Classification
- Retinoic acid signaling regulates development of the dorsal forebrain midline and the choroid plexus in the chick
- New findings for phenotype-genotype correlations in a large European series of holoprosencephaly cases
- Septopreoptic Holoprosencephaly: A Mild Subtype Associated with Midline Craniofacial Anomalies
- The Cavum Septi Pellucidi: Why Is It Important?
- Mutations in the BMP pathway in mice support the existence of two molecular classes of holoprosencephaly
- Central Roles of the Roof Plate in Telencephalic Development and Holoprosencephaly
- Middle interhemispheric variant of holoprosencephaly: A very mild clinical case
- Facial hemangioma and cerebral corticovascular dysplasia: A syndrome associated with epilepsy
- MRI shows abnormal white matter maturation in classical holoprosencephaly
- Middle interhemispheric variant of holoprosencephaly: A distinct cliniconeuroradiologic subtype
- Neuroanatomy of holoprosencephaly as predictor of function: Beyond the face predicting the brain