
Abstract
Summary: Krabbe's disease is an autosomal recessive leukodystrophy with well-documented intracranial findings on both CT scans and MR images. We herein present what is thought to be the second case of Krabbe's disease with spinal involvement shown on MR images as abnormal contrast enhancement of the lumbosacral nerve roots. The typical intracranial findings of T2 hyperintensity without contrast enhancement were present within the periventricular white matter, but there was no area of abnormal signal intensity or enhancement within the substance of the spinal cord. We briefly review the pathophysiology, clinical presentation, and imaging findings of Krabbe's disease. Spinal abnormalities may precede the onset of brain abnormalities, and MR imaging may be a useful diagnostic tool in cases of Krabbe's disease and other leukodystrophies.
Krabbe's disease, also known as globoid cell leukodystrophy, is an autosomal recessive leukodystrophy with an incidence estimated at 1 in 100,000 to 200,000 live births (1). Although the intracranial imaging findings of Krabbe's disease have been well documented, we are reporting what is thought to be the second case with spinal abnormalities shown on MR images (the first such case with coexisting intracranial abnormalities). In addition, we briefly review the clinical, laboratory, and imaging findings of Krabbe's disease.
Case Report
A 16-month-old male patient was admitted to the hospital for evaluation of a possible neurodegenerative process. The patient was the product of an uncomplicated term delivery, and he had initially achieved normal developmental milestones. At approximately 13 months, the patient began experiencing a gradual regression in motor skills to the point at which he could no longer stand or sit upright without assistance. Additionally, he was experiencing low-grade fevers, episodes of vomiting and diarrhea, decreased oral intake, and increasing irritability. The results of a physical examination were remarkable for an increase in muscle tone, lower extremity spasticity, and episodic flexion of the upper and extension of the lower extremities (exacerbated by stimulation). The patient has no siblings, and his family history was unremarkable.
MR imaging revealed symmetric areas of T2 prolongation within the periventricular white matter, centered in the region of the centrum semiovale (Figs 1 and 2). There was no intracranial area of abnormal enhancement after the IV administration of contrast material (Fig 3). Imaging of the spine displayed abnormal contrast enhancement of the lumbosacral nerve roots (Fig 4A and B) but no area of abnormal signal intensity or enhancement within the substance of the spinal cord (Fig 5A and B). Routine laboratory and CSF analyses were remarkable for an elevated CSF protein of 84 mg/dL (normal, 15–45 mg/dL). Lysosomal enzyme testing revealed a galactocerebroside beta-galactosidase level that was 3 SD below the normal, confirming the diagnosis of Krabbe's disease. Follow-up of this patient has been of short duration, but the symptoms have progressed with increasing spasticity. No follow-up imaging has been performed.

Axial view T2-weighted MR image (4200/90 [TR/TE]) reveals symmetric areas of T2 prolongation (arrows) within the centrum semiovale
fig 2. Coronal view fluid-attenuated inversion recovery image (8002/137.5) reveals symmetric areas of abnormal signal intensity (arrows) within the periventricular white matter
fig 3. Coronal view T1-weighted image (400/20) obtained after the IV administration of contrast material reveals no enhancement within the periventricular white matter (arrows)
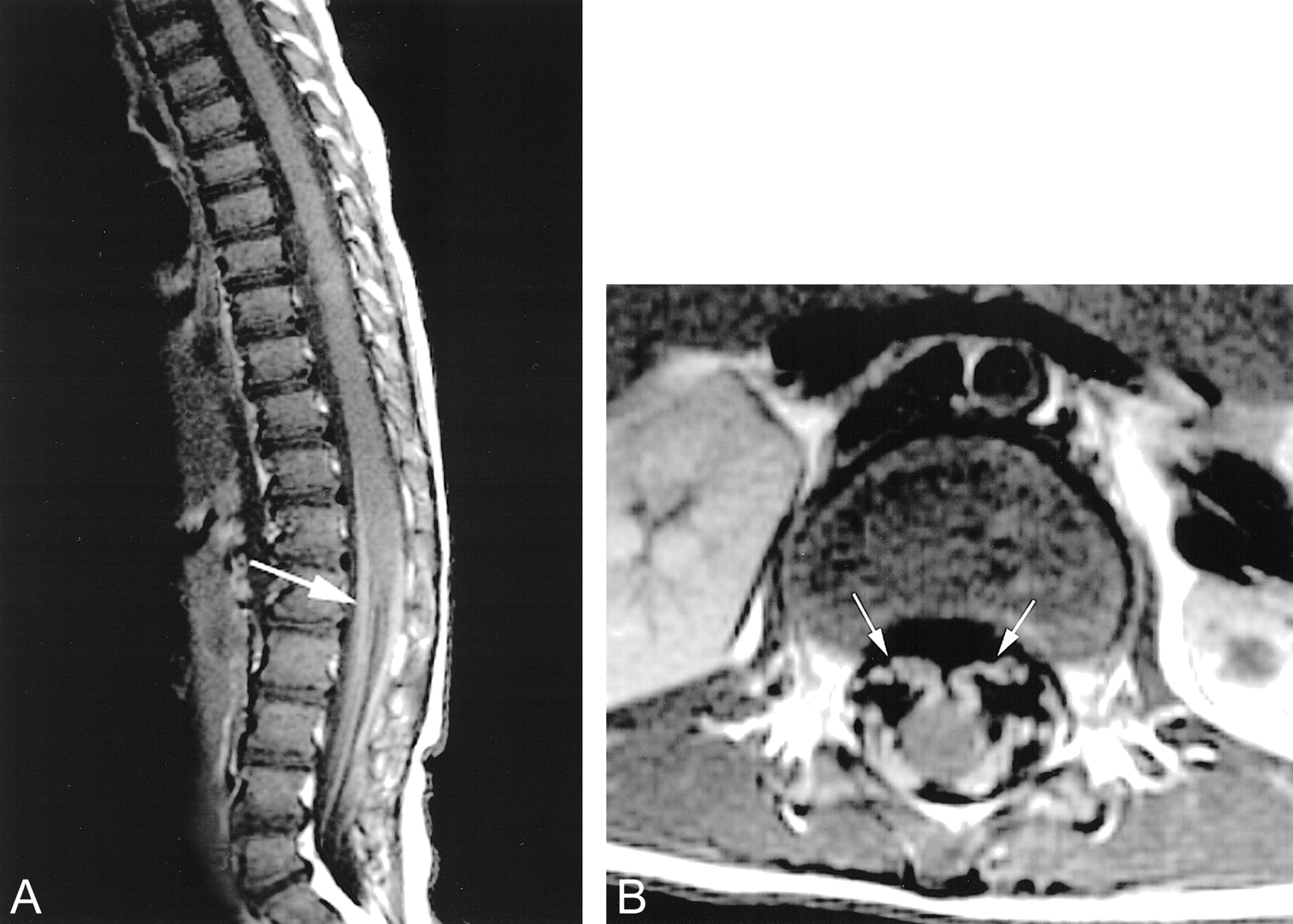
T1-weighted MR images (450/20) of the lumbosacral region reveal striking contrast enhancement of the lumbosacral nerve roots (arrows).
A, Sagittal view.
B, Axial view.

T2-weighted MR images (4000/105) display normal signal intensity within the substance of the spinal cord and lumbosacral nerve roots.
A, Sagittal view.
B, Axial view.
Discussion
Krabbe's disease is one of the more common leukodystrophies, inherited in an autosomal recessive fashion. It is caused by a deficiency in the lysosomal enzyme galactocerebroside beta-galactosidase, with the genetic locus mapped to chromosome 14 (2). Galactocerebroside beta-galactosidase is responsible for the conversion of galactosylceramide to ceramide and psychosine to sphingosine. There are alternative enzymes that can break down galactosylceramide when galactocerebroside beta-galactosidase is deficient. However, this is not the case for psychosine (3). Psychosine accumulates at toxic levels in Krabbe's disease, resulting in oligodendrocyte death and impaired Schwann cell function (4, 5). The normal harmonious relationship between myelin breakdown and production is therefore disturbed, causing a severe demyelination involving both the central and peripheral nervous system (6). Histologically, there is myelin destruction, gliosis, and the presence of the characteristic multinucleated “globoid” cells (macrophages) within the perivascular regions of the affected white matter (7).
In Krabbe's original publication (8), he described involvement of the spinal cord with “destruction of the medullary sheaths,” but he made no mention of the lumbosacral nerve roots in his autopsy series. Proposed mechanisms for the abnormal nerve root enhancement have included altered vascular permeability with breakdown of the blood-nerve barrier as a result of perivascular inflammation or infiltration (6, 9) and enhancement in areas of active myelin breakdown (10). Animal models inflicted with galactocerebroside beta-galactosidase deficiency have shown breakdown of the blood-nerve barrier with preservation of the blood-brain barrier (9, 11, 12), accounting for the selective lumbosacral nerve root enhancement seen in our case. With the exception of a few reported cases (13), contrast enhancement is not associated with the intracranial areas of myelin destruction.
The clinical significance of the lumbosacral nerve root enhancement remains uncertain; our patient displayed symptoms of upper motor neuron abnormality (ie, spasticity) attributable to the intracranial abnormalities. The clinical presentation may prove beneficial in distinguishing the spinal imaging findings associated with Krabbe's disease from those associated with other pathologic abnormalities that possess a similar appearance on MR images but present with lower motor neuron symptoms, such as Guillain-Barré syndrome (14).
Krabbe's disease is classically divided into three subgroups: early infantile, late infantile, and juvenile forms (1). Others have simplified the classification into “infantile” and “late onset” (15, 16). A full discussion of the various subtypes is beyond the scope of this case report, but a few key points are worth mentioning. The infantile form is the most common subtype and generally presents with progressive irritability and then stiffness, vision loss, and rapid motor or mental decline in a child who had previously achieved normal developmental milestones (6, 15). This subtype is rapidly progressive, with death usually occurring by 2 years of age (15). The late onset variety typically presents after 10 years of age and has a more protracted clinical course. The late onset type has been known to mimic a peripheral neuropathy, and the disease in these patients often goes undiagnosed for many years (15).
Diagnosis is usually suspected on the basis of clinical and radiologic findings, with confirmation by lysosomal enzyme testing revealing a deficiency of galactocerebroside beta-galactosidase. Early in the course of the disease, nonenhanced CT examinations may show areas of symmetrically increased attenuation within the basal ganglia, thalami, and centrum semiovale (3, 16, 17). The pathogenesis of this increased attenuation remains debated, with hypotheses suggesting areas of calcification (18) and grouping of the globoid cells (17). These areas of increased attenuation may actually precede any identifiable white matter abnormalities on either CT scans or MR images (17). The middle stages of the disease are characterized by areas of hypoattenuation on CT scans and T2 prolongation on MR images within the periventricular white matter (centrum semiovale). Progressive cerebral and cerebellar atrophy herald the late stages of the disease. The cranial nerves may be affected, with multiple cases of thickened optic nerves noted at both autopsy (8) and MR imaging (3). Bernal and Lenn (10) reported a case in which there was abnormal enhancement of multiple cranial nerves.
To the best of our knowledge, there has been only one previously reported case of the spinal involvement of Krabbe's disease with imaging (6). Vasconcellos and Smith (6) presented a case of abnormal enhancement involving the lumbosacral nerve roots in a 7-month-old child with an infantile form of Krabbe's disease. Their case had no intracranial manifestations of the disease revealed by either CT or MR imaging at the time of presentation or by follow-up MR imaging performed 1 week later. Because their patient had the classic clinical presentation of Krabbe's disease, it is unlikely that this represented a variation in the enzyme defect that preferentially affected the peripheral nervous system. Instead, it seems more plausible that the spinal involvement preceded any intracranial imaging abnormalities. Our case shares the similar finding of striking enhancement of the lumbosacral nerve roots but differs in that the typical cerebral white matter abnormalities are present. Although it seems that the lumbosacral nerve root enhancement may precede the intracranial abnormalities, this will require future investigation with larger case series to evaluate the usefulness of spinal MR imaging of patients with suspected leukodystrophy. To our knowledge, lumbosacral manifestations (if any) of other leukodystrophies have not been reported. Further investigation will also be required to determine whether all the sub-types of Krabbe's disease show similar changes in the lumbosacral region or whether this finding is limited to the infantile forms.
Conclusion
Krabbe's disease is one of the more common leukodystrophies with well-documented intracranial findings. However, spinal manifestations on MR images are only recently being reported. Lumbosacral nerve root contrast enhancement may accompany or possibly precede the intracranial findings of Krabbe's disease, and spinal imaging may prove useful in diagnosing or differentiating the various leukodystrophies. Krabbe's disease should be included in the differential of enhancing lumbosacral nerve roots in infants and children, along with other such entities as Guillain-Barré syndrome (14), lymphoma/leukemia, Dejerine-Sottas disease (19), and metastatic disease.
Footnotes
1 Address reprint requests to Curtis A. Given II, MD, Department of Neuroradiology, Wake Forest University Baptist Medical Center, 2nd Floor, Meads Hall, Medical Center Drive, Winston-Salem, NC 27157.
References
- Received March 27, 2001.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}