
Abstract
Summary: We present the first report of a patient with atypical hemifacial microsomia (HFM) and unilateral aplasia of the floor of the middle cranial fossa, glenoid fossa, and portions of her posterior fossa. The patient also developed a Chiari I malformation with cervical syrinx over a 3-year interval. This case report highlights the critical role of imaging in revealing serious, but clinically occult, structural abnormalities, as well as the evolution in the pathogenetic understanding of HFM.
Hemifacial microsomia (HFM) is a complex malformation syndrome with varied nomenclature throughout the literature, a large host of genetic and teratogenic associations, and a wide spectrum of clinical features involving the facial skeleton and other organ systems (1). HFM, at a minimum, involves abnormal development of the first branchial arch (2). A widely accepted theory of its pathogenesis centers on abnormal stapedial artery development (3). Likely, a genetic predisposition to abnormal vascular development, combined with extrinsic factors, leads to bleeding at the anastomosis of the developing stapedial artery (4). The resulting combination of ischemia and hematoma leads to abnormal formation of the first branchial arch derivatives during weeks 3 through 5 of embryogenesis. The patient we describe, however, had what we believe is a unique presentation within the malformation complex, with complete unilateral aplasia of the floor of her middle cranial fossa, portions of her posterior cranial fossa, and a small, but otherwise normal, ipsilateral mandible.
Case Report
A 14-year-old girl presented with torticollis of unclear etiology. The torticollis was not present at birth, but had increased over the previous three years. Initial workup at another institute, including CT and MR imaging of her cervical spine 3 years prior, concluded that she had hypoplasia of the left occiput-C1 junction, and no further treatment was recommended. The patient also complained of mild left facial asymmetry. Although she noted occasional headaches that worsened with bending over or running, she did not complain of neck pain or paresthesias. She was athletically active, participating in both soccer and drill team events at her school.
On physical examination, she was noted to have mild head tilt to the left. She had facial asymmetry, with the left mandible and maxilla appearing smaller than the right. She had mild left microtia, with an abnormal upper pinna and posterior rotation of the auricle. In addition, the skin anterior to her left ear demonstrated dermal hypoplasia, reduction in soft-tissue bulk, and overlying vascular hyperplasia. This lesion was prone to bleeding with minor trauma. Her cranial and motor neurologic examination was normal. There was minimal patchy loss of sensation over her shoulders, but this was difficult to reproduce.
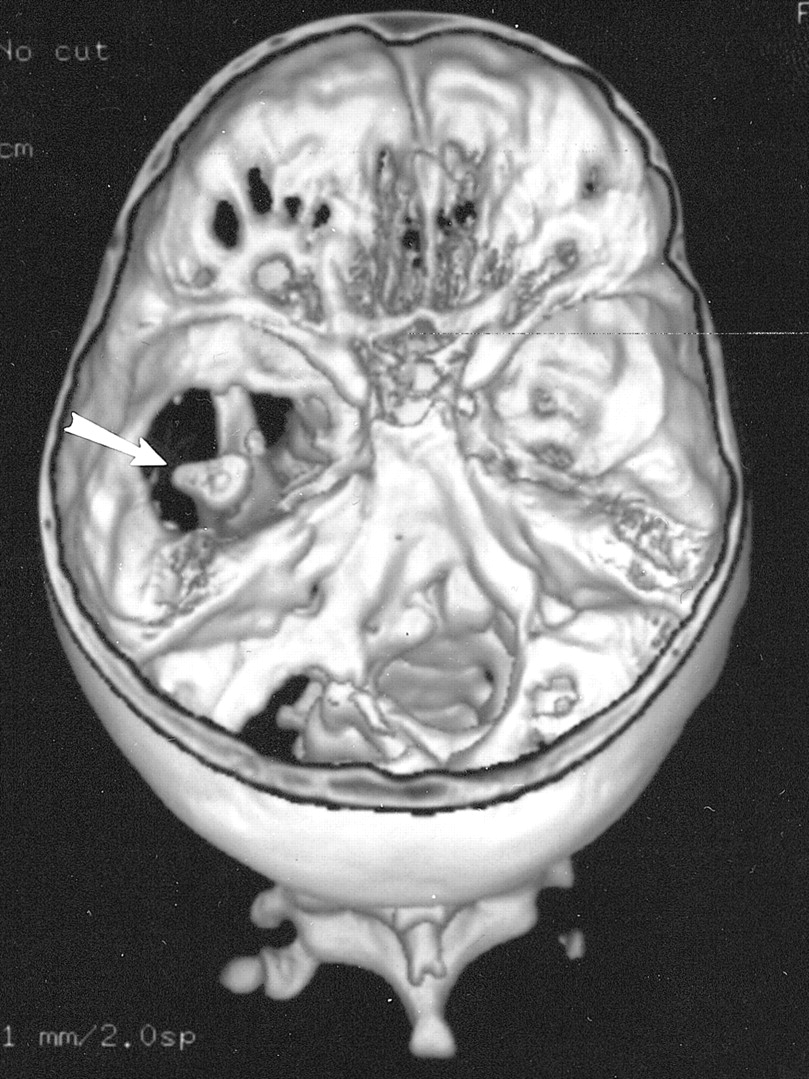
A CT examination of her neck and head was ordered to better evaluate her torticollis and left facial asymmetry. This revealed complete aplasia of the floor of her left middle cranial fossa, as well as portions of the posterior fossa floor (Fig 1). The small but otherwise normally formed left mandible and condyle articulated with the base of her temporal lobe (Fig 2A). Also noted was mild hypoplasia of her left maxilla and zygomatic body. The temporal portion of the zygomatic arch was absent. The inner and middle ear were normally formed, with normal ossicles. The styloid process and hyoid bone were normal. MR angiography revealed normal formation and course of the major intracerebral vessels.

Three-dimensional CT reconstruction of the skull base seen from above shows large defects in the base of the left middle and posterior cranial fossa. The left mandibular condyle is seen through the bony defect (arrow)
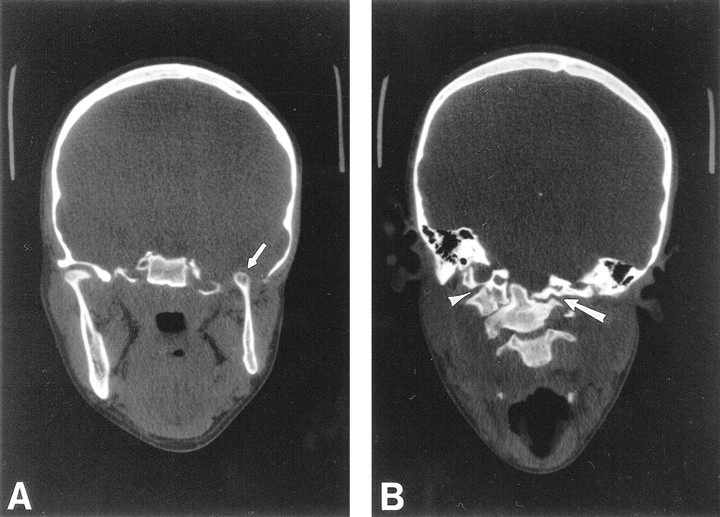
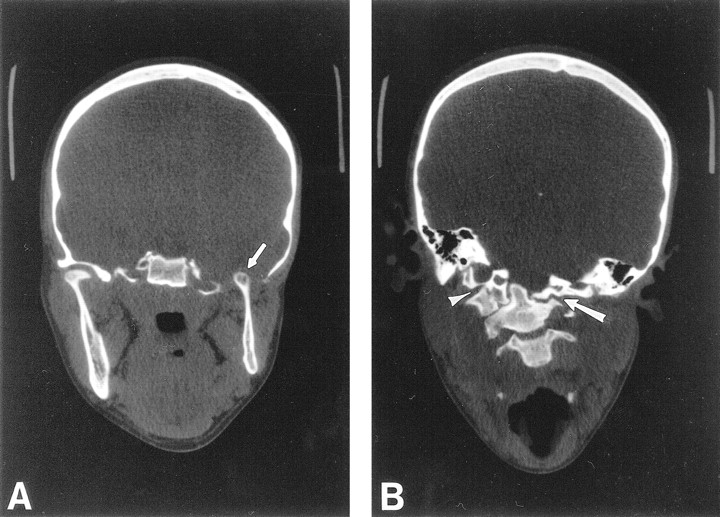
Coronal CT through the madibular condyles and craniocervical junction.
A, Coronal CT through the mandibular condyles shows a normal right temporomandibular joint, and aplasia of the floor of the left middle cranial fossa and glenoid fossa. The left mandibular ramus and condyle are small but otherwise normal. The left mandibular condyle projects into the middle cranial fossa (arrow) and articulates with the base of the temporal lobe.
B, Coronal CT through the craniocervical junction shows absence of the left arch of C1, with the lateral mass of C2 articulating with the skull base (arrow). The right arch of C1 is fused to the occipital condyle (arrowhead), resulting in a focal right convex curve at the craniocervical junction.
CT of her neck revealed an aplastic left arch of C1, with the lateral mass of C2 and the dens articulating with the skull base (Fig 2B). The right arch of C1 was fused to the occipital condyle. As a result, the patient had a right convex scoliosis at the craniocervical junction. A follow-up MR examination of her cervical spine, to better evaluate the spinal anatomy, revealed a Chiari I malformation with 17 mm of tonsilar descent (Fig 3), and a syrinx at the C2–C3 level, with cord edema extending caudally to the C5–C6 level. The tonsilar ectopia and syrinx were not present on the outside study performed 3 years previously (not shown). Appropriate surgical treatment was initiated.

T1-weighted 550/10/2 (TR/TE/excitations) sagittal MR image through the craniocervical junction shows a Chiari I malformation with a 17-mm cerebellar tonsilar descent and a syrinx at the C2–C3 level (arrow)
Discussion
HFM is a malformation complex that involves, at a minimum, asymmetrical abnormal development of the first branchial arch derivatives (2). Initially, the term hemifacial microsomia was limited to abnormal aural, oral, and mandibular development, with the Goldenhar syndrome reserved for patients with the addition of epibulbar dermoids, vertebral, and cardiac anomalies (1). As this malformation complex has evolved, with extensive phenotypic overlap and association with abnormalities of other organ systems, including cardiovascular, pulmonary, and genitourinary, this distinction has lost validity. The etiology is heterogeneous, with many genetic and teratogenic associations (1).
A widely accepted theory for the pathogenesis of HFM is that hemorrhage associated with the formation of the stapedial arterial system during embryogenesis disrupts normal development of the first, and also second, arch derivatives (3). The evolution of this theory is instructive. It was initially based on an understanding of the embryonic development of the stapedial artery. Subsequently, a teratogen-induced and a genetically engineered animal phenocopy of HFM have been created; both reveal hematomas of the stapedial artery as the first pathologic event.
McKenzie (2), building on earlier embryologic observations, postulated that abnormal development of the stapedial artery could explain the constellation of abnormalities in HFM, or what he called the first arch syndrome. During embryonic weeks 3 to 5, the blood supply to the first arch depends on the successive relay of three vessels: the first aortic arch, then the stapedial artery (a second arch derivative), and finally the definitive blood supply from the external carotid artery. He reasoned that any disruption of this sequence, which needed to be precisely timed and coordinated, could result in ischemia to embryonic tissue during a critical period of rapid growth and differentiation.
Poswillo (3) was successful in creating a phenocopy of what he called the first and second branchial arch syndrome in the offspring of mice and monkeys administered the teratogens triazene and thalidomide, respectively. Microscopic examination of affected embryos revealed hematomas surrounding the anastomoses of the developing stapedial artery. Poswillo hypothesized that variations in the size and shape of the hematoma would explain the phenotypic variability; the larger the hematoma, the more structures would be involved and the longer the time for the hematoma to resorb, and thus the less time for the embryonic tissue to repair and redifferentiate.
Most recently, investigators have created a transgenic mouse line with an insertional mutation that results in a phenocopy of HFM, including microtia and malocclusion (5). The mutation was the deletion of a 23 kb segment on mouse chromosome 10, which the investigators have designated Hfm, for hemifacial microsomia-associated locus. These genetically engineered mice also showed rupture of the stapedial vasculature as the first pathologic event, lending further credence to the theory of stapedial artery rupture and resultant hematoma and ischemia first formulated nearly four decades earlier. It seems likely that the cervical vertebral anomalies long associated with HFM also have, as their fundamental pathogenesis, abnormalities of the early vasculature. This would be in keeping with experiments showing the relationship between congenital vertebral abnormalities of formation and segmentation with abnormal intersegmental artery development (6).
Addressing the specific malformations in our patient, all of the affected facial bones are derivatives of the first branchial arch. In particular, the mandible, maxilla, zygomatic bone, and squamous temporal bone (forming the floor of the middle cranial fossa and glenoid fossa) all form from direct ossification of first branchial arch dermal mesenchyme (7). The abnormalities of the upper pinna and mild microtia are also consistent; the three auricular hillocks of the first arch give rise to the helix, as well as the tragus and cymba concha (7). Multiple case reports have detailed abnormal formation or absence of the mandibular condyle (8), but the complete absence of the glenoid fossa and floor of the middle cranial fossa, with a small but otherwise normal mandibular condyle, is unique.
The occipital bone is derived from the chondrocranium, a cartilaginous precursor. The defects in the posterior cranial fossa in our patient are likely related to her vertebral anomalies, in that both the chondrocranium and vertebral bodies are derived from sclerotomes, or somite-derived mesodermal cells. In particular, the base of the occipital bone arises from a combination of the occipital sclerotome and the first cervical sclerotome, and is thus a modified vertebral element (7).
This patient's abnormal temporomandibular joint also highlights some of the unique features of this joint. Unlike other joints, the temporomandibular joint is formed from two distinct blastema that grow toward each other (9). An opposing theory by investigators that the joint arose from a single mesenchymal condensation that differentiated into a temporal and mandibular component has been disproved by embryologic studies (9). The unique morphology of the temporomandibular joint in our patient, consisting of a small, but otherwise normal, mandibular condyle, and complete aplasia of the glenoid fossa, is strong proof of the two blastema model.
The findings of cerebellar tonsilar ectopia with syrinx and cord edema were not present on an MR examination obtained 3 years before, and are likely due to the patient's abnormal craniocervical junction. Several theories have been advanced as to how obstruction of the foramen magnum by tonsilar ectopia might lead to a syrinx and cord edema (10), with most involving increased spinal CSF pressure driving CSF across the spinal parenchyma into the central canal. Ours is only the second report of a syrinx associated with HFM (11). The patient in the prior report had upper cervical anomalies and the presence of cerebellar tonsils at the foramen magnum level. A syrinx was diagnosed by a metrizamide myelogram and delayed CT scanning. MR imaging was not performed.
In conclusion, to our knowledge, this is the first report of a patient with atypical hemifacial microsomia with complete aplasia of the floor of the middle cranial fossa and glenoid fossa. This is only the second case report of a cervical syrinx associated with the HFM complex. This case highlights the critical value of imaging in diagnosing two serious, but clinically occult, structural anomalies of the skull and spine. In particular, the absence of intervening bone between the mandibular condyle and the temporal lobe was cause for serious concern in a young patient involved in contact sports such as soccer. Likely, the good occlusion between her maxilla and mandible was protective, but clearly any trauma to the mandible could have lead to brain injury (12). Despite her unique anatomy, in aggregate, her malformation remains consistent with developmental abnormalities restricted to derivatives of the first branchial arch. The normal formation of her styloid and hyoid bones reveals that her second branchial arch was not affected.
The history of the attempt to understand HFM is also noteworthy. The stapedial artery theory was based on embryologic observations of the complex succession of arteries supplying the first branchial arch. Subsequently, a teratogen-created animal model of HFM with stapedial artery hematomas was created, supporting the initial hypothesis. Most currently, a molecular developmental model has been created, demonstrating identical pathogenesis. The critical locus on mouse chromosome 10 has been identified, and investigators are trying to identify the specific gene or genes involved and their mechanisms. As a unifying hypothesis, it seems possible that a genetic predisposition to vascular anomalies, coupled with various extrinsic insults, might lead to hemorrhage at critical points in embryonic development. Depending on the location of the vascular event, this could lead to abnormal development of the craniofacial and vertebral skeleton, and other organ systems as well. The unique constellation of findings in our patient may further the understanding of the pathogenesis of related disorders.
Footnotes
1 Address reprint requests to Raymond W. Sze, Department of Radiology, Children's Hospital and Regional Medical Center, 4800 Sand Point Way NE, CH-69, Seattle, WA 98105.
R.W. Sze is supported by the American Roentgen Ray Society Scholarship.
References
- Received September 26, 2000.
- Accepted after revision March 17, 2001.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.