
Abstract
BACKGROUND AND PURPOSE: Chordoid glioma is a new clinicopathologic entity that occurs in the region of the hypothalamus/anterior third ventricle. The aims of this study were to describe the characteristic radiographic features of chordoid glioma, identify specific imaging features that may enable differentiation of chordoid glioma from other suprasellar tumors, and increase neuroradiologists' awareness of this newly described tumor, facilitating prospective diagnosis.
METHODS: CT scans and/or MR images of six patients with chordoid glioma were reviewed retrospectively to determine whether any characteristic radiographic features would emerge. Reports of the clinical presentation, pathologic findings, and radiographic findings of another six patients were reviewed and included, for a total patient population of 12 (mean age ± SD, 46 ± 13 years).
RESULTS: Imaging features were strikingly similar for all tumors. In each case, the mass was ovoid, was well circumscribed, was located in the region of the hypothalamus/anterior third ventricle, and enhanced uniformly and intensely. Tumors were hyperdense to gray matter on CT scans and were isointense on T1-weighted MR images and slightly hyperintense on long-TR MR images. In two patients, vasogenic edema extended into the optic tracts, and in three, there was hydrocephalus.
CONCLUSION: Chordoid glioma is a recently described unique histopathologic entity that has been added to the World Health Organization glioma classification scheme and must be included in the differential diagnosis of a suprasellar mass. Distinctive imaging features are its location, ovoid shape, hyperdensity on CT scans, and uniform intense contrast enhancement.
Brat et al (1) recently reported a series of low-grade neoplasms arising in the region of the anterior third ventricle/hypothalamus and determined that they represent a unique histopathologic entity. This tumor was named chordoid glioma because of its distinctive histologic appearance, reminiscent of chordoma, and its avid staining with glial fibrillary acidic protein. An evaluation of the imaging studies of patients who were diagnosed with chordoid glioma revealed a striking similarity among all cases. The distinctive imaging features are described herein, and the potential for prospective radiologic diagnosis of chordoid glioma are discussed.
Methods
Patients
Imaging studies (CT and/or MR imaging) of four patients who presented with chordoid glioma after March 1998 and two who presented before that time (Table, patients 1–6) and were included in the initial report of this entity (1) were reviewed retrospectively by two authors (M.G.P., T.J.P.). Reports of the clinical presentations, pathologic findings, and radiographic findings of another six patients were also reviewed and included, for a total patient population of 12 (three male and nine female patients; mean age ± SD, 46 ± 13 years). All diagnoses were determined by histopathologic examination according to published methods at the time of tumor resection (1).
Patient characteristics and imaging findings
Imaging Protocol
CT (patients 2, 5, 7, and 12) was performed in the axial plane, before and after administration of a bolus IV injection of iohexol (2 mL/kg), with contrast-enhanced coronal reformatting performed for one patient (patient 2). MR imaging was performed on a 1.5-T system in all cases. Although the MR imaging studies were conducted at several institutions, comparable, standard MR parameters were used, with approximate TR as follows: spin-echo T1-weighted sagittal (500/15 [TR/TE]) and double-echo T2-weighted axial (2500/15/100 [TR/TE/TE]). For two patients (patients 3 and 5), a fluid-attenuated inversion recovery sequence (8912/142; inversion time, 2200 ms) was also obtained. All patients received IV administered gadopentetate dimeglumine (0.2 mL/kg), and contrast-enhanced T1-weighted images were subsequently obtained in the sagittal, axial, and/or coronal planes. Tumors were manually traced using MedVol 1.7 (Bruce Hall, MD, Johns Hopkins University) to measure tumor volumes from the four patients (patients 1, 3, 5, and 6) who had MR images that contained an internal scale.
Image Review
Tumor location, size, shape, margins, MR signal characteristics (on T1-, T2-, or proton density–weighted sequences), where applicable, and enhancement pattern or other characteristics, including involvement of adjacent structures, extent of vasogenic edema, and presence or absence of hydrocephalus, were noted.
Pathology
Each patient underwent a subtotal resection through a frontal craniotomy approach, except for patients 7 and 12 who underwent total resections and patient 3 who underwent a biopsy. Surgical specimens were formalin-fixed, routinely processed, sectioned at 4 μm, and stained with hematoxylin and eosin. For immunohistochemical studies, sections were deparaffinized and subjected to antigen retrieval by either limited protein digestion or steaming (20 min at 80o C). Slides were then incubated at room temperature with antibodies directed toward glial fibrillary acidic protein (polyclonal, 1:6000; Dako Corp., Carpenteria, CA) and epithelial membrane antigen (monoclonal, 1:2000; Dako). Antibodies were detected using the avidin-biotin complex method, using diaminobenzidine as the chromogen.
Results
Imaging features were strikingly similar for all tumors (Table). In each case, the mass was located in the region of the hypothalamus and anterior third ventricle, was ovoid in shape, and was well circumscribed (Fig 1). Mean tumor volume (±SD) was 16.6 ± 13.9 cm3 for patients 1, 3, 5, and 6. Hydrocephalus was noted in three patients (patients 7, 9, and 10) but in none who were available for retrospective analysis. All tumors were separate from the pituitary gland. On CT scans, tumors were hyperdense to gray matter with homogeneous enhancement (Fig 2). Tumors were isointense on T1-weighted MR images and isointense to slightly hyperintense on long TR MR images, and all enhanced essentially uniformly (Fig 3). The tumors of patients 1, 8, and 9 contained a central cystic or necrotic area (Fig 4), although the corresponding biopsy specimens did not indicate the presence of necrosis. In two of the patients available for review (patients 3 and 5), T2 bright signal due to vasogenic edema extended into the optic tracts (Fig 3).

Representative appearance of chordoid glioma on MR image. Note the location of the mass in the hypothalamus/third ventricle region, dense enhancement, and greatest dimension in the superoinferior orientation.
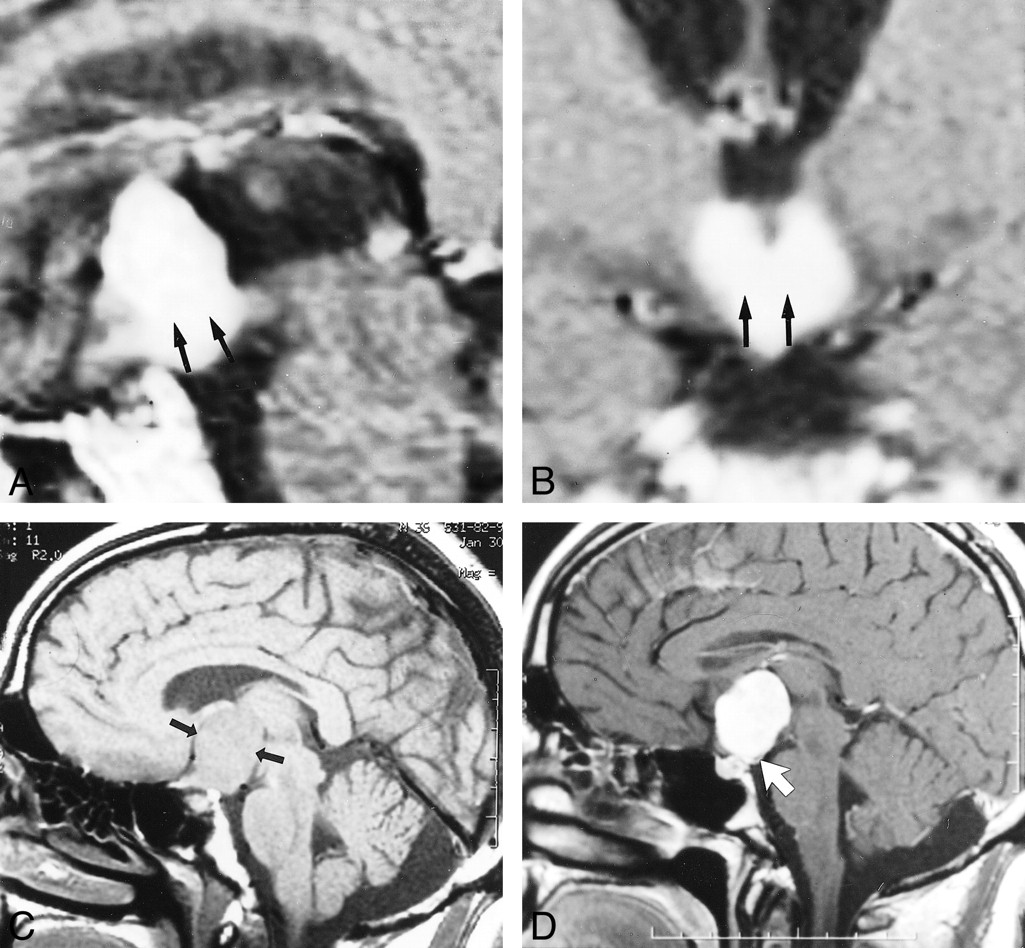
A, Sagittal contrast-enhanced spin-echo MR image (500/15) of Patient 1, a 59-year-old man. Note hypothalamic involvement (arrow).
B, Coronal contrast-enhanced spin-echo MR image (500/15) of Patient 1, a 59-year-old man. Note hypothalamic involvement (arrow).
C, Unenhanced sagittal spin-echo MR image (500/14) of Patient 3, a 36-year-old man. Arrows demarcate the suprasellar mass.
D, Contrast-enhanced sagittal spin-echo MR image (500/14) of Patient 3, a 36-year-old man. Note the posterior displacement of the infundibulum (arrow).

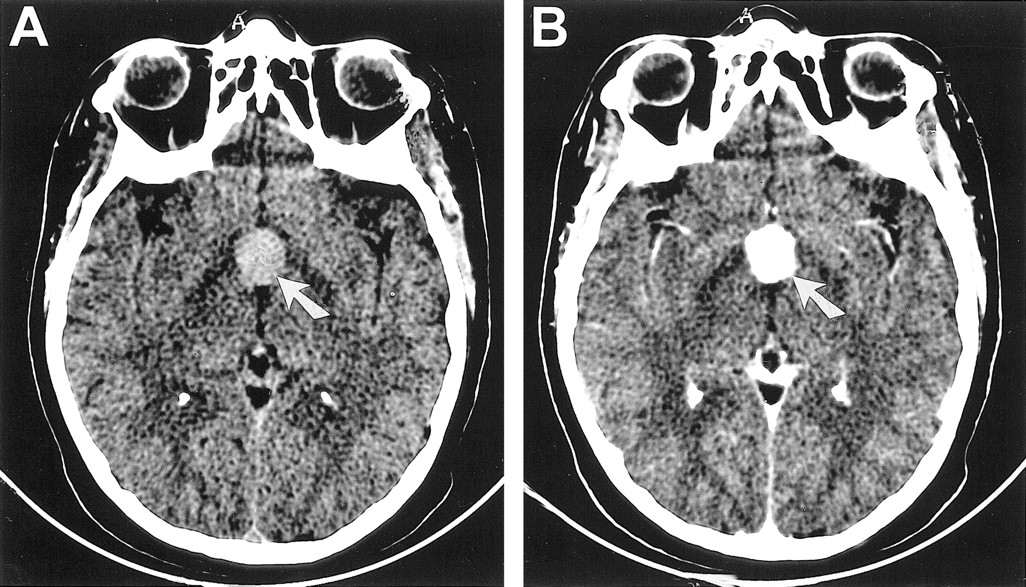
CT scans of Patient 2, a 41-year-old woman with chordoid glioma.
A, Unenhanced axial CT scan reveals a hyperdense mass (arrow) that enhances uniformly in the suprasellar region.
B, Contrast-enhanced axial CT scan reveals a hyperdense mass that enhances uniformly (arrow) in the suprasellar region.
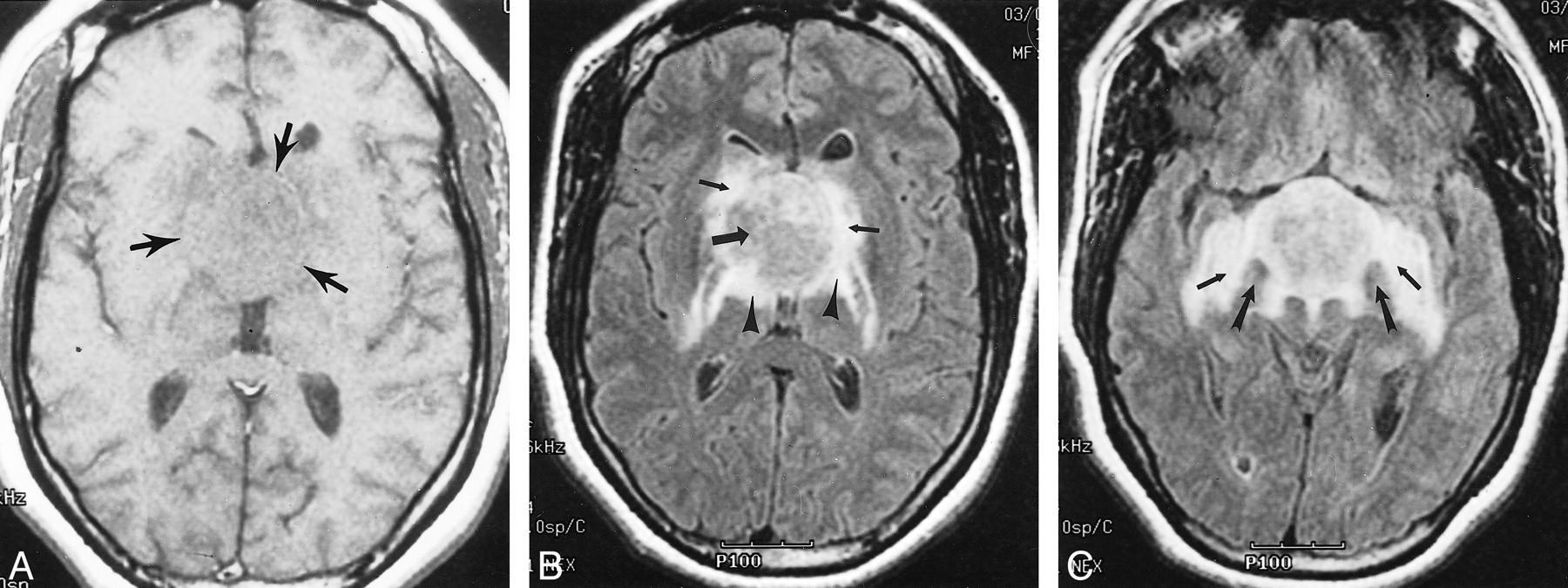
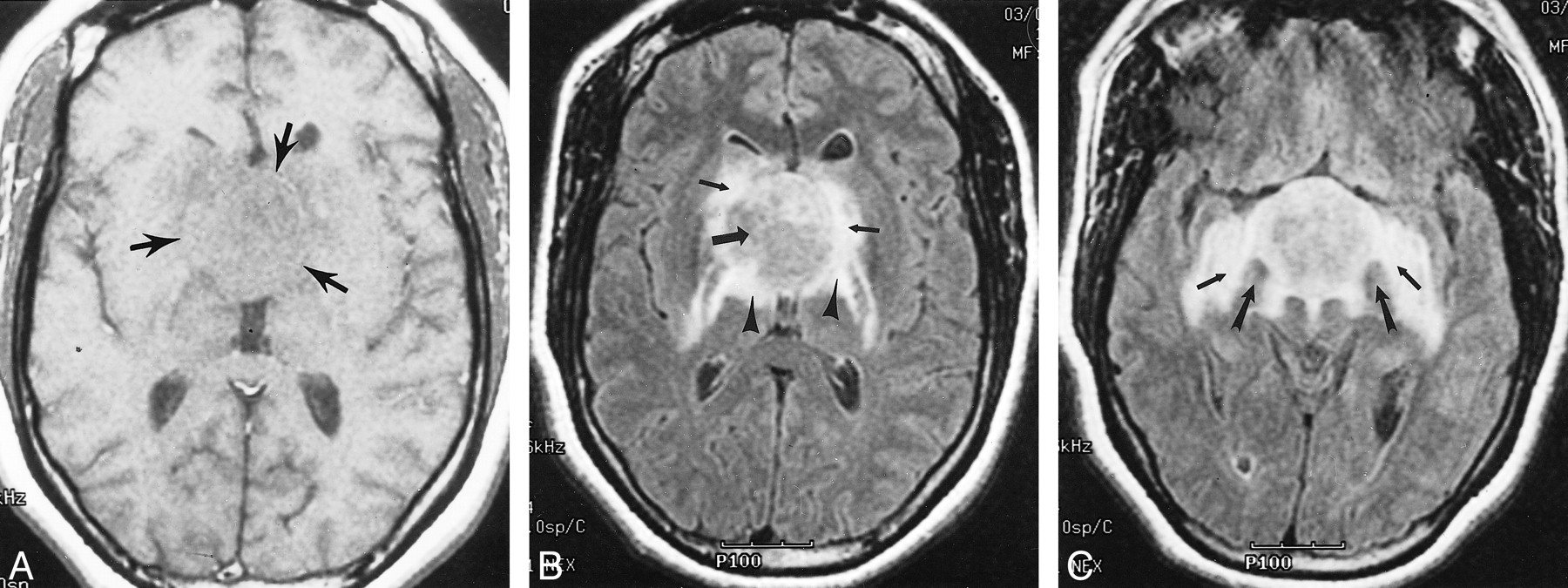
Images of Patient 5, a 35-year-old woman with chordoid glioma.
A, Axial T1-weighted spin-echo MR image (450/20) reveals an isointense hypothalamic/third ventricular mass (arrows).
B, Axial fluid-attenuated inversion recovery MR image (8912/142; inversion time, 2200 ms) reveals an iso- to slightly hyperintense hypothalamic/third ventricular mass (large arrow). Note vasogenic edema within the basal ganglia (small arrows), posterior limbs of the internal capsules, and lateral geniculate ganglia of the thalami (arrowheads).
C, Axial fluid-attenuated inversion recovery MR image (8912/142; inversion time, 2200 ms) obtained at a level approximately 1.5 cm inferior to that shown in B. Note splaying of the cerebral peduncles (large arrows) and bilateral involvement of the optic tracts (small arrows).
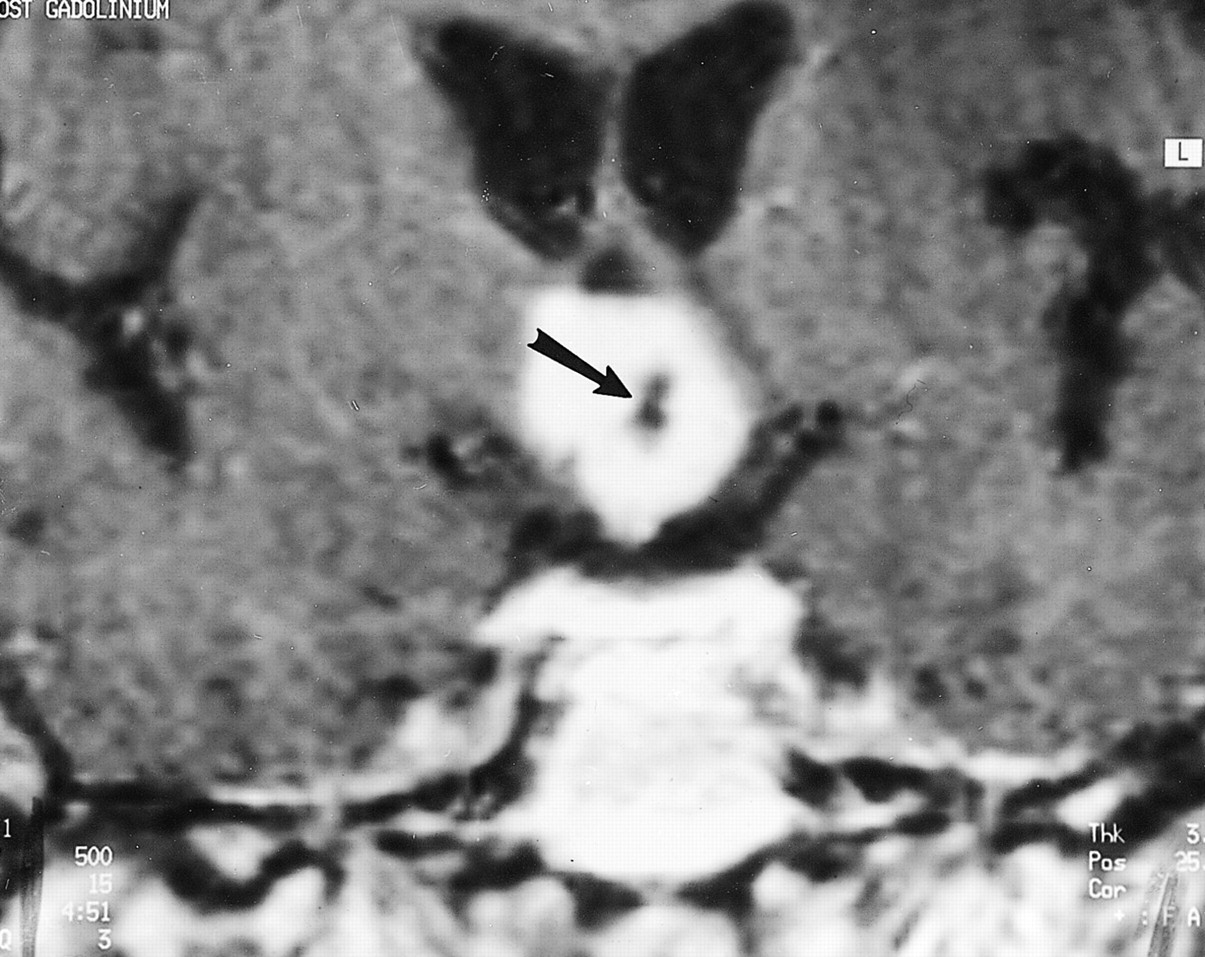
Image of Patient 1, a 59-year-old man with chordoid glioma. Contrast-enhanced coronal T1-weighted spin-echo MR image (500/15) reveals a densely enhancing hypothalamic/third ventricular mass with a central nonenhancing component representing a small cyst or necrosis (arrow)
Detailed pathologic results have been described elsewhere but generally revealed clusters and cords of oval-to-polygonal epithelioid cells with abundant eosinophilic cytoplasm (Fig 5) (1). No anaplastic features were identified, and there was no appreciable infiltration of the surrounding brain parenchyma. The overall cytologic features were consistent with those of a low-grade neoplasm.
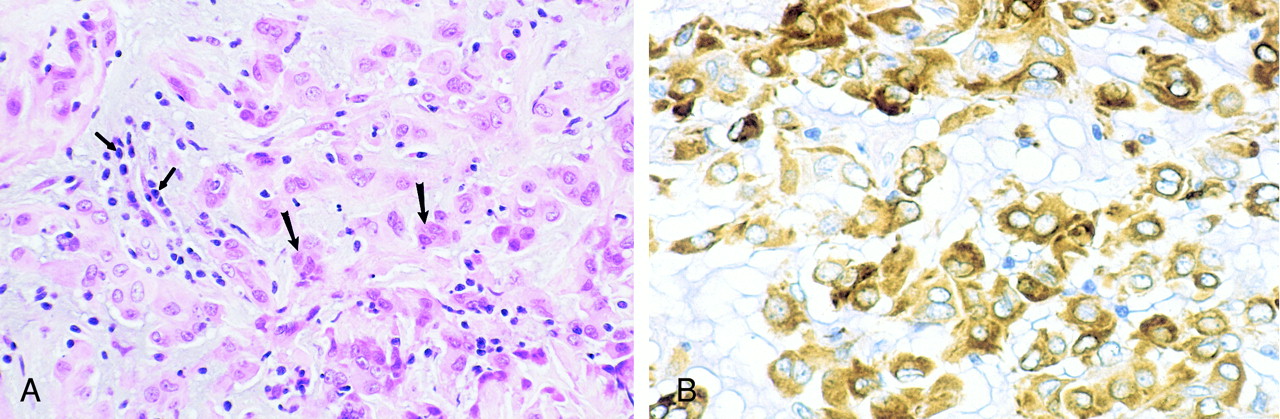
Photomicrographs of chordoid glioma (Patient 5).
A, Hematoxylin and eosin stain of chordoid glioma reveals cords and clusters of eosinophilic epithelioid tumor cells (large arrows) dispersed within a slightly bluish mucinous matrix. A lymphoplasmacytic infiltrate is consistently present (small arrows).
B, Glial fibrillary acidic protein stain of chordoid glioma tumor cells shows strong, diffuse, cytoplasmic immunoreactivity.
Discussion
During the past decade, a series of histopathologically perplexing neoplasms, each arising in the region of the hypothalamus and anterior third ventricle, were seen in consultation at our institution and at the Mayo Clinic. These neoplasms had the histologic appearance of cords and clusters of epithelioid cells within a mucinous background along with a low-grade lymphoplasmacytic infiltrate, reminiscent of a chordoma or chordoid meningioma (Fig 5). Unlike chordomas or chordoid meningiomas, however, which are not glial in origin, these tumors all stained avidly for the glial cell marker glial fibrillary acidic protein. This new tumor did not conform to any existing glioma histopathologic classification system, leading Brat et al (1) to propose that chordoid glioma was a separate pathologic entity. Since that original report, four new cases have come to our attention and at least six other cases have appeared in the literature (2, 3). Interestingly, in 1995, Wanschitz et al (4) reported a case of a peculiar suprasellar meningioma that exhibited atypical expression of glial fibrillary acidic protein and probably represents another example of a chordoid glioma. Chordoid glioma has recently been added to the World Health Organization glioma classification scheme (5). Although the incidence of chordoid glioma cannot yet be evaluated because of its recent description, it seems to be an uncommon tumor.
The cell of origin for chordoid glioma remains unknown. A recent case report indicates that the neoplasm arises from the subependymal tissue (6). That same report noted that despite the glial nature of these lesions, the presence of histiocytes and giant cells suggests the presence of other cellular types with unknown roles and significance. Another strong possibility is that the tumors arise from the hypothalamus. That is supported in that most lesions are tightly adherent to the hypothalamus at surgery, resulting in a difficult resection, although the tumor is otherwise well circumscribed.
Patients presented clinically in a variety of fashions, generally related to the local mass effect of the tumor (eg, headaches, obstructive hydrocephalus, homonymous hemianopsia, and hypothalamic dysfunction) (Table). The current treatment of choice for chordoid glioma is surgical resection. As mentioned above, even though the tumor is otherwise well marginated, surgeons have frequently reported that the mass is tightly adherent to the hypothalamus, resulting in either subtotal resection or hypothalamic symptoms (eg, diabetes insipidus resulting from aggressive removal). Tumors that have undergone subtotal resection tend to recur. Chemotherapy and radiation therapy have been unsuccessful in the few cases in which these modes of therapy have been applied. Despite that chordoid glioma is a pathologically low-grade tumor, prognosis in the limited number of cases has been relatively poor, primarily because of its location and the difficulty in obtaining complete surgical resection without the patient's suffering severe hypothalamic symptoms.
Because the local anatomy is complex and composed of tissues that may develop into tumors, such as the cranial nerves, the cavernous sinuses, the pituitary gland, and the skull base, 80% of suprasellar lesions are tumors (7, 8). That there are many possible tumor types is a diagnostic dilemma for the radiologist who discovers a suprasellar mass. Pituitary macroadenomas, craniopharyngiomas, meningiomas, metastases, optic and hypothalamic pilocytic astrocytomas, and aneurysms account for more than 75% of these lesions. Other less common entities include Rathke cleft cysts, hamartomas of the tuber cinereum, and granulomatous diseases such as sarcoid, tuberculosis, and eosinophilic granuloma. Tumors of the third ventricle constitute an uncommon subset of suprasellar masses. These tumors have been addressed previously from both radiologic and pathologic perspectives (9–12). Tumors involving the third ventricle can arise either intrinsically (eg, ependymoma, choroid plexus papilloma/carcinoma, or colloid cyst) or extrinsically with extension into the third ventricle (eg, astrocytoma, germinoma, or pineal region masses). Chordoid glioma further expands the differential diagnosis of third ventricular masses.
Despite that only six of the 12 cases presented herein were available for review, we think that the six cases contain all of the critical features of chordoid glioma detectable on cross-sectional images. On CT scans, chordoid glioma could be confused with other hyperdense lesions that may appear in the region of the hypothalamus/third ventricle, such as lymphoma, meningioma, and aneurysm; its dense uniform enhancement is reminiscent of each of those entities. MR imaging is superior for diagnosing this tumor because it clearly depicts the hypothalamic involvement by enabling multiplanar imaging. Sagittal images clearly depict the infundibulum to be displaced posteriorly (Fig 1) and the greatest dimension of these ovoid masses to be generally in the superoinferior orientation. By contrast, Rathke cleft cysts and tuber cinereum hamartomas generally displace the infundibulum anteriorly (7). The relative T2 signal isointensity of chordoid glioma is again reminiscent of lymphoma or meningioma; however, the bilaterally symmetrical posterior extension of vasogenic edema (eg, into the optic tracts and lateral geniculate ganglia of the thalami) (Fig 3) may help to distinguish this tumor from the latter two entities. Differentiation of chordoid glioma from the optic/hypothalamic glioma may be difficult using imaging alone. That the latter is a tumor of childhood enables differentiation on clinical grounds. On MR images, chordoid glioma is better circumscribed than is the optic/hypothalamic glioma and does not extend into the optic chiasm or optic tracts; however, edema from mass effect, rather than enhancing tumor tissue, may be evident in the optic tracts (Fig 3). Hydrocephalus is a variable feature that is more dependent on the precise location of the mass than on its overall size. For example, patient 9 had a relatively small tumor with hydrocephalus (Table) whereas patient 5 (Fig 3), with a significantly larger mass, did not. Consideration of these specific imaging findings may enable the prospective diagnosis of chordoid glioma by neuroradiologists.
Conclusion
Chordoid glioma is a recently described, unique pathologic entity that has been added to the World Health Organization glioma classification scheme and must be considered in the differential diagnosis of third ventricular/hypothalamic tumors. Distinctive imaging characteristics are its consistent location, ovoid shape, hyperdensity on CT scans, and uniform intense contrast enhancement. When present, mass effect causing vasogenic edema tends to be bilaterally symmetrical and may involve the optic tracts. When confronted with a suprasellar mass with these imaging features, the neuroradiologist must consider chordoid glioma in the differential diagnosis.
Footnotes
1 Presented at the 37th Annual Meeting of the American Society of Neuroradiology, San Diego, April 1999.
↵2 Address reprint requests to Martin G. Pomper, MD, PhD, Division of Neuroradiology, Johns Hopkins University School of Medicine, 600 N. Wolfe Street, Baltimore, MD 21287.
- Received February 7, 2000.
- Copyright © American Society of Neuroradiology




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}