
Abstract
Summary: We describe the MR imaging findings in a case of granulomatous hypophysitis due to Wegener's granulomatosis. A high index of suspicion of hypophysitis based on imaging findings allowed successful medical management and helped avoid surgery. The MR imaging features included a thickened stalk, a diffusely and uniformly enlarged gland, a normal size or minimally enlarged sella, and enhancement of the optic chiasm.
Hypophysitis is of two types: lymphocytic and granulomatous. Granulomatous hypophysitis is rare, with an estimated incidence of one case per 10 million per year (1). It may be associated with systemic conditions, such as Takayasu's disease (1), ruptured intrasellar Rathke's cleft cyst (2, 3), and Crohn's disease (4) and with thyroiditis and lymphocytic adrenalitis (5). A few cases of Wegener's granulomatosis with granulomatous hypophysitis have also been reported (6–9). We describe a case of Wegener's granulomatosis with granulomatous hypophysitis and suggest possible indications revealed by MR imaging to allow medical management and prevent surgery.
Case Report
A 48-year-old woman with a known diagnosis of Wegener's granulomatosis presented with a 2-week history of progressive visual loss and gradually worsening polyuria, polydipsia, lethargy, and headaches. Notably, there was no history of galactorrhea or menstrual abnormalities and there were no signs or symptoms of anterior pituitary gland dysfunction except for mild hypothyroidism. The patient's visual acuity was reduced to 0.8 in both eyes, with concentric reduction in visual fields. There was evidence of mild papilledema bilaterally. Laboratory investigations revealed mildly low thyroid stimulating hormone, dilute urine, and an erythrocyte sedimentation rate of 60 mm.
The patient had a many-year history of systemic Wegener's granulomatosis affecting her respiratory and urinary systems. The diagnosis of Wegener's granulomatosis had been made on the basis of a nasal biopsy and a strongly positive c-anti-neutrophil cytoplasmic antibody. The systemic manifestations of disease were well controlled at the time of the current presentation. The patient was not receiving any regular medication.
An MR examination was performed to evaluate the cause of the visual loss and diabetes insipidus. MR imaging revealed a sellar mass with suprasellar extension, which was hypointense on T1-weighted images, was hyperintense on T2-weighted images, and showed heterogeneous enhancement on contrast-enhanced images (Fig 1). The optic chiasm was elevated as a consequence of the mass. The optic chiasm showed enhancement on the contrast-enhanced images (Fig 1, arrows). There was also heterogeneous soft tissue in the sphenoid sinus. The mucosa in the sinus was thickened and enhanced, whereas the contents of the sinus were typical of inspissated fluid. On the basis of the imaging characteristics of the pituitary lesion, the differential diagnosis included an aggressive neoplasm, sphenoid sinus infection with extension into the pituitary gland, and hypophysitis. Although granulomatous hypophysitis is very rare, in view of the patient's history of Wegener's granulomatosis and the imaging features (suprasellar extension, enhancement of the optic chiasm), it was decided to administer a trial of steroids rather than to subject the patient to biopsy and/or resection of the lesion. A regimen of low-dose thyroxine and desmopressin acetate was also begun.
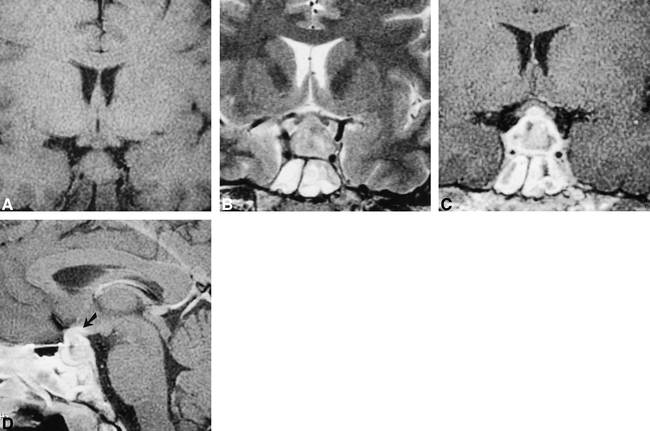
MR images obtained through the pituitary gland show diffuse enlargement of the pituitary gland. The gland shows diffuse heterogeneous enhancement that extends to the optic chiasm (arrow). Also note the presence of mucosal thickening in the bilateral sphenoid sinus.
A, T1-weighted coronal (450/15/4 [TR/TE/excitations]) MR image.
B, T2-weighted coronal (4000/90/3; echo train length, 8) MR image.
C, Contrast-enhanced T1-weighted coronal (450/15/4) MR image.
D, Contrast-enhanced T1-weight sagittal (450/15/4) MR image.
The patient showed a dramatic response to steroids, achieving significant improvement in her vision and visual field during the next 2 weeks. A follow-up MR examination 3 weeks later showed nearly complete resolution of the mass, although there continued to be prominence of the pituitary infundibulum with enhancement (Fig 2). There was no evidence of recurrence during 18 months of follow-up. The patient continued to receive desmopressin acetate and achieved normal thyroid function.
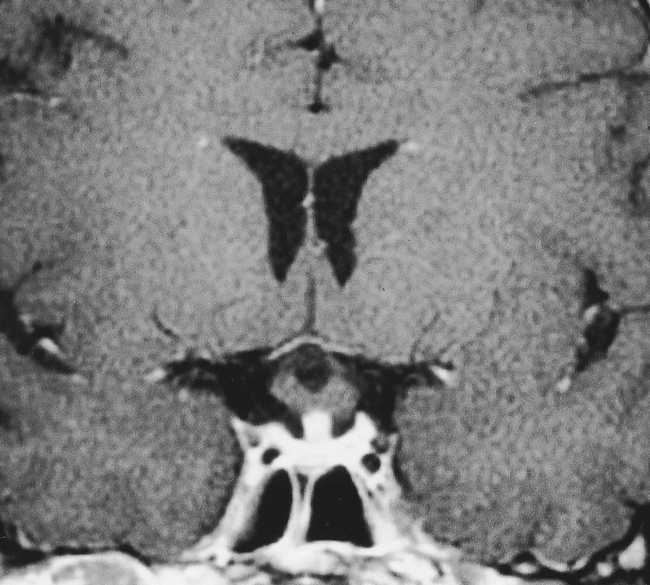
Follow-up MR examination after 6-week administration of steroids. Contrast-enhanced T1-weighted coronal (450/15/4) MR image shows dramatic resolution in the size of the pituitary gland. No enhancement is seen in the optic chiasm
Discussion
Histologically, hypophysitis is broadly divided into two categories: lymphocytic and granulomatous (10). Lymphocytic hypophysitis is better described, with more than 60 cases reported in the literature (10). It is thought to have an autoimmune pathogenesis (11). More than 30% of patients have other coexistent autoimmune disorders (12, 13). An autoimmune pathogenesis is also suggested by immunological characterization and ultrastructural evidence showing that cytotoxic lymphocytes intimately contact anterior pituitary cells (14, 15). Additionally, lymphocytic hypophysitis can be induced in rats by injection of isologous and homologous pituitary tissue (16). Histologically, lymphocytic hypophysitis is characterized by infiltration of the anterior pituitary gland with lymphocytes and plasma cells and by fibrosis, which finally leads to destruction of the gland (12, 13). The majority of cases reported in the literature have occurred in female patients during pregnancy or soon after delivery (10).
The description of granulomatous hypophysitis is limited to few case reports and case series (1–10, 17–26). It is characterized by granulomas with epithelioid histiocytes and multinucleated giant cells. Lymphocyte collections also occur but are not the dominant feature (17, 18). It was initially thought to be a distinct entity occurring in isolation, but a few cases have recently been described as occurring in association with other systemic disorders, such as Takayasu's disease (1), ruptured intrasellar Rathke's cleft cyst (2, 3), Crohn's disease (4), and with thyroiditis and lymphocytic adrenalitis (5). A few cases of Wegener's granulomatosis with granulomatous hypophysitis have also been reported (6–9). Because of their ultrastructural similarity and autoimmune origin for both granulomatous and lymphocytic hypophysitis, it has been suggested that the two entities may have similar pathogenetic backgrounds and may even represent different stages of the same disease (20). The coexistence of granulomatous and lymphocytic hypophysitis has also been described as occurring in a single patient (27).
Approximately 20% of the cases of Wegener's granulomatosis include CNS involvement (6), most commonly presenting as cranial neuropathies (6, 7). Subarachnoid and/or intraparenchymal hemorrhage due to vasculitis has also been described to occur in these patients (6). Involvement of the pituitary gland is distinctly unusual and most often presents with diabetes insipidus (6–9). However, presentation with visual symptoms and anterior pituitary dysfunction has been reported previously (7). The majority of patients reported in the literature on Wegener's granulomatosis with pituitary involvement have had other evidence of activity of disease, including sinusitis, lung nodules, nephropathy, cutaneous vasculitis, and arthritis (6). The absence of these in our patient at the time of the present episode is distinctly unusual.
The clinical presentation of granulomatous hypophysitis is either hypopituitarism, diabetes insipidus, hyperprolactinemia, meningitis, visual disturbance due to mass effect, or a combination of the above (6–8, 10, 22). Involvement of cranial nerves and presentation with ophthalmoplegia has also been described as occurring in one case (24). The clinical presentation in our case with headache and visual disturbance with a relatively normal hypothalamic-pituitary axis is unusual.
Honegger et al (10) observed that the most striking feature of lymphocytic and granulomatous hypophysitis is the increased CSF cell count and related meningitis. Although our patient did not undergo a CSF examination, there was no clinical evidence of meningitis revealed by repeated clinical examination.
The importance of a preoperative diagnosis is obvious. A significant proportion of patients would respond to medical management, such as steroids. Surgery is unlikely to succeed in the complete removal of the affected tissue and is probably more likely to result in long-standing hypopituitarism. The presence of a coexistent granulomatous disease, such as Wegener's granulomatosis, as in our case, definitely raises the index of suspicion toward this condition. Additional imaging features that have been described to favor the diagnosis of hypophysitis include a thickened stalk, a diffusely enlarged gland, a normal size or minimally enlarged sella, and extension of enhancement to the optic chiasm (10, 20, 26). The presence of an inflammatory reaction in the sphenoid sinus, as was observed in our patient, has been described earlier (10). Although coincidental sinus disease cannot be excluded, spread of the inflammation from the pituitary fossa seems plausible (10). All these imaging features were present in our case, allowing us to consider the possibility of hypophysitis with reasonable certainty.
Some authors have advocated the routine use of surgery early in the course of the disease to make an accurate diagnosis and to remove the diseased tissue (10). A high rate of recurrence associated with the use of steroids has also been reported (10, 15). A case unresponsive to steroids has also been reported (28). We think that the approach to this condition should be individualized. The coexistence of a systemic condition with typical imaging findings, as in this case, may not need to be subjected to surgery but can be treated with steroids under close supervision. The dramatic clinical and imaging response in our case, lack of long-term hypopituitarism, and no evidence of recurrence during an 18-month follow-up period justify our management.
To conclude, we present an unusual case of Wegener's granulomatosis with hypophysitis, which showed excellent response to treatment with steroids. The presence of the systemic disease and characteristic MR imaging features, such as a diffusely enlarged pituitary gland, a relatively normal sized sella, a thickened stalk, and enhancement of the adjacent optic chiasm, allowed the correct preoperative diagnosis.
Footnotes
↵1 Address reprint requests to Walter Kucharczyk, MD, FRCPC, Department of Medical Imaging, University of Toronto, 150 College Street, Toronto, Ontario M5S 3E2, Canada.
References
- Received January 3, 2000.
- Copyright © American Society of Neuroradiology




{kind=link}
{kind=link}