
Abstract
BACKGROUND AND PURPOSE: The purpose of this study was to describe the cranial MR imaging manifestations of osteopetrosis. These features have not previously been reported in the literature.
METHODS: Cranial MR studies, obtained with a uniform imaging protocol, were reviewed in 47 patients with osteopetrosis. Thirty-four patients had autosomal recessive (malignant) osteopetrosis (AROP), seven had intermediate osteopetrosis (IOP), and six had either type I or type II autosomal dominant osteopetrosis (ADOP I or II). The prevalence of abnormalities was tabulated and compared with the specific osteopetrosis variants.
RESULTS: All patients with osteopetrosis had thickening and sclerosis of the calvaria. Ventriculomegaly, tonsillar herniation, proptosis, and dural venous sinus stenosis were observed in the majority of patients with AROP and ADOP I. Optic nerve sheath dilatation occurred in many of the patients with AROP and in all patients with ADOP I. Acquired cephaloceles were also observed only in these two groups. Optic nerve atrophy and optic canal stenosis were observed in a majority of patients with AROP, IOP, and ADOP II. Middle ear fluid was prevalent in AROP and IOP, present in over half the patients in each group. Features seen most prevalently, or exclusively, in AROP included stenosis of the internal carotid and vertebral arteries and extramedullary hematopoiesis.
CONCLUSION: The cranial MR imaging features of osteopetrosis are both shared and unique among the various subtypes of the disease. The specific cranial and intracranial manifestations reflect the predominant calvarial or skull base patterns of bone thickening. The unique features seen in patients with AROP probably reflect the early age of onset and the greater severity of this form of the disease.
Osteopetrosis most likely results from more than one genetic or biochemical defect, with at least five types of the disease having been described. The two most commonly seen forms are autosomal recessive (malignant) osteopetrosis (AROP) and autosomal dominant (benign) osteopetrosis (ADOP). AROP is associated with deficient osteoclastic resorption of the primary spongiosa. This form of osteopetrosis usually has its onset in infancy. Patients exhibit poor osseous growth and remodeling, and anemia, infection, and hemorrhage complicate obliteration of the marrow spaces (1). Other manifestations of AROP include cranial nerve palsies, optic atrophy, and stenoses of the petrous internal carotid artery (ICA), the jugular veins within narrowed jugular foramina, and the cervical vertebral arteries within narrowed transverse foramina (2–4). A more uncommon form of the disease, intermediate osteopetrosis (IOP), also demonstrates autosomal recessive inheritance. However, this variant tends to present later in childhood than the more malignant form. Typically, these patients have short stature and experience some of the more aggressive features of malignant osteopetrosis (1, 5). Frequently, IOP is distinguished from the malignant form only when a milder clinical course evolves with age.
ADOP presents in adulthood, although in children from families with osteopetrosis it may be identified earlier. The adult type may be asymptomatic and discovered incidentally on plain radiographs, as when fractures occur. A small percentage of patients with ADOP experience more severe complications of the disease, including facial nerve palsy, visual loss, deafness, bone pain, and osteomyelitis. Two types of ADOP have been differentiated on the basis of clinical and radiographic criteria. Type I ADOP is characterized by diffuse calvarial sclerosis and thickening with diffuse sclerosis of bone in the spine and pelvis. Histologically, few abnormalities are apparent in the modeling of trabecular bone. Indeed, the bones are quite strong and fractures are rare. In type II ADOP, calvaria are less severely involved, but the skull base is sclerotic and thickened (6). Other radiographic features include hyperostotic vertebral endplates (the “rugger-jersey spine”) and endobones (or a “bone-within-a-bone appearance”), the latter signifying a reduced ability of the osteoclasts to model and remodel the bone, leading to a markedly increased risk of fracture (7).
Although the cranial CT features of AROP have been described (8), no detailed study has been undertaken of the cranial MR imaging manifestations in the broader spectrum of osteopetrosis. We report the cranial MR imaging findings on examinations performed in a large number of patients with a variety of osteopetrotic conditions.
Methods
The study objectives and scan protocol were approved by our institutional review board. Cranial MR imaging examinations of 47 patients with osteopetrosis were evaluated, and the findings classified according to type of osteopetrosis on the basis of the criteria described below.
History
Patients were categorized by age at presentation, symptoms, and family history. AROP was determined from evidence of leukoerythroblastic anemia, splenomegaly, and/or some degree of cranial nerve compromise manifesting before the age of 6 months.
Histomorphometric Data
All patients underwent a bone biopsy, and those with osteopetrosis were identified by the finding of cartilaginous islands within bone, the histologic equivalent of endobones. These islands represent unresorbed cartilage remaining from the cartilaginous primordia of the bones.
Radiographic Data
All patients underwent a skeletal survey; those with osteopetrosis had evidence of bone sclerosis throughout the skeleton. Other sclerosing bone dysplasias and diseases are not associated with sclerosis in both the axial and appendicular skeleton. A secondary radiographic finding in osteopetrosis is the presence of endobones. Disease manifesting in adulthood was subdivided into types I or II on the basis of patterns of skull and spinal involvement and clinical and laboratory findings.
Laboratory Data
Disease with adult onset was further classified by levels of creatine phosphokinase brain-band isoenzyme, which were high in patients with the adult form, type II.
Based on these features, 34 patients (19 male and 15 female, aged 2 weeks to 29 years) had AROP, or malignant osteopetrosis. Seven patients (six male and one female, aged 2 to 17 years) had IOP. Six patients (aged 19 to 48 years) had ADOP, three with type I (two male and one female) and three with type II (all female). The MR imaging protocol consisted of T1-weighted sagittal spin-echo images and T2-weighted axial fast spin-echo (FSE) images (all 47 patients), intracranial 3D time-of-flight (3D-TOF) MR angiography and 2D-TOF coronal MR venography (46/47 patients), and axial cervical 2D-TOF MR angiography (42/47 patients). All patients with osteopetrosis seen at our institution during a 7-year period were imaged. The abnormalities observed on review of these MR studies were tabulated retrospectively.
Results
All patients with osteopetrosis had both thickening and sclerosis (markedly hypointense signal within bone on all MR imaging sequences) of the calvaria (see Table and Fig 1). Calvarial thickening was further rated as mild (estimated to be between one and two times normal calvarial thickness), moderate (two to three times normal calvarial thickness), or severe (more than three times normal calvarial thickness). The severity of calvarial thickening was greatest in AROP (14 of 33 were rated as severe, seven as moderate) and ADOP I (all three were rated as severe). All cases of calvarial thickening in ADOP II were rated as moderate. Calvarial thickening was rated as mild in five of seven cases of IOP, and as moderate in two. Scalloping of the inner table was seen most frequently in ADOP I (Fig 2A).
Cranial MR imaging findings, by type of osteopetrosis
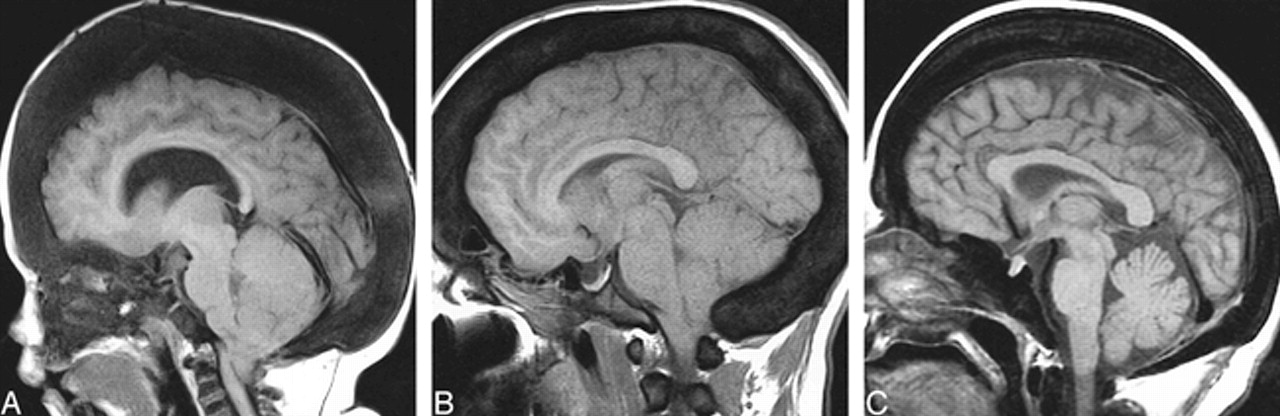
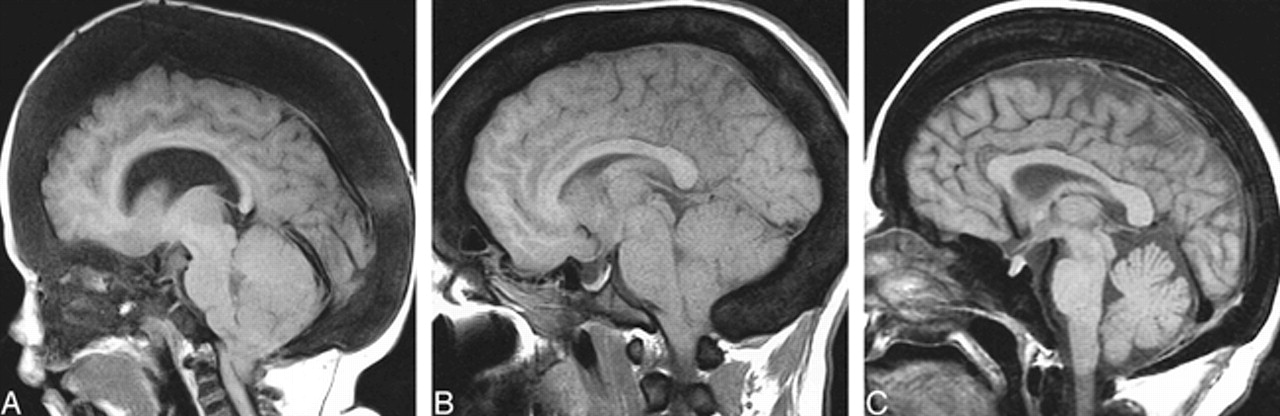
Calvarial thickening.
A, AROP. Sagittal T1-weighted MR image shows thickening of the calvaria and facial bones. Note hypointensity of skull and cervical vertebra, and cerebellar tonsillar ectopia.
B, ADOP I. Sagittal T1-weighted image shows calvarial thickening and hypointensity, as well as tonsillar ectopia.
C, ADOP II. Sagittal T1-weighted image shows only a moderately thickened calvaria; however, the skull base is more significantly thickened than in the example of ADOP I (B). Compare the small sellar volume with that in ADOP I (B).

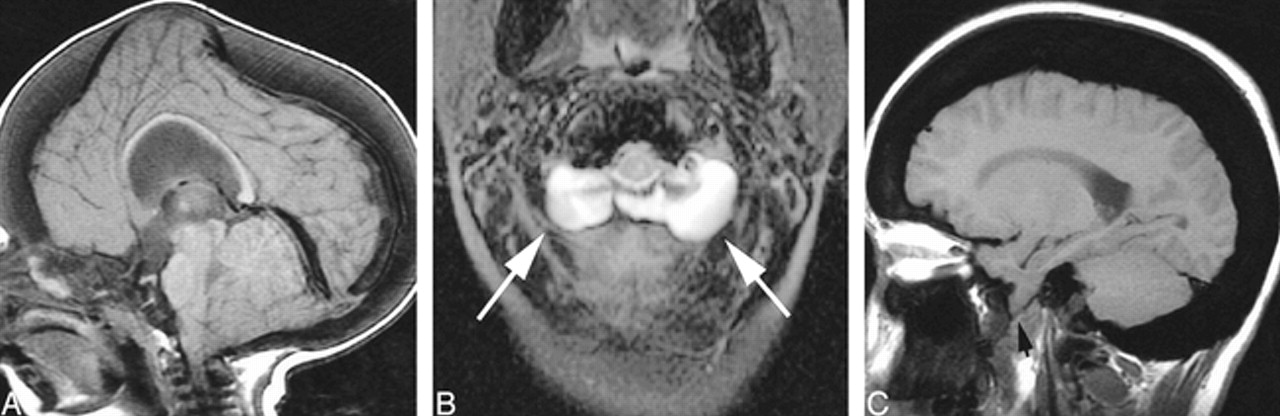
Optic nerve/sheath manifestations.
A, Prominent optic nerve sheath dilatation, ADOP I. Axial T2-weighted FSE image shows scalloped inner table. The optic canals are not stenotic.
B, AROP. Axial T2-weighted FSE image shows optic canal stenosis and optic nerve atrophy.
C, IOP. Axial T2-weighted FSE image shows optic canal stenosis and optic nerve atrophy, with more severe atrophy evident on the right in this case.
Ventriculomegaly, tonsillar herniation (Fig 1), proptosis, and dural venous sinus stenosis (Fig 3) occurred in the majority of patients with AROP and ADOP I. Optic nerve sheath dilatation (Fig 2A) occurred in many of the patients with AROP and in all the patients with ADOP I. Cephaloceles, either spontaneous (Fig 4) or postsurgical (Fig 5), were also observed only in these two groups, though in neither group was there a high prevalence of cephaloceles. Despite the frequency of venous stenosis in patients with osteopetrosis (particularly AROP and ADOP I), venous infarcts were not observed in any patients. Optic nerve atrophy and optic canal stenosis were present in a majority of patients with AROP, IOP, and ADOP II (Fig 2B and C). Middle ear fluid was prevalent in AROP and IOP, present in over half the patients in each group.
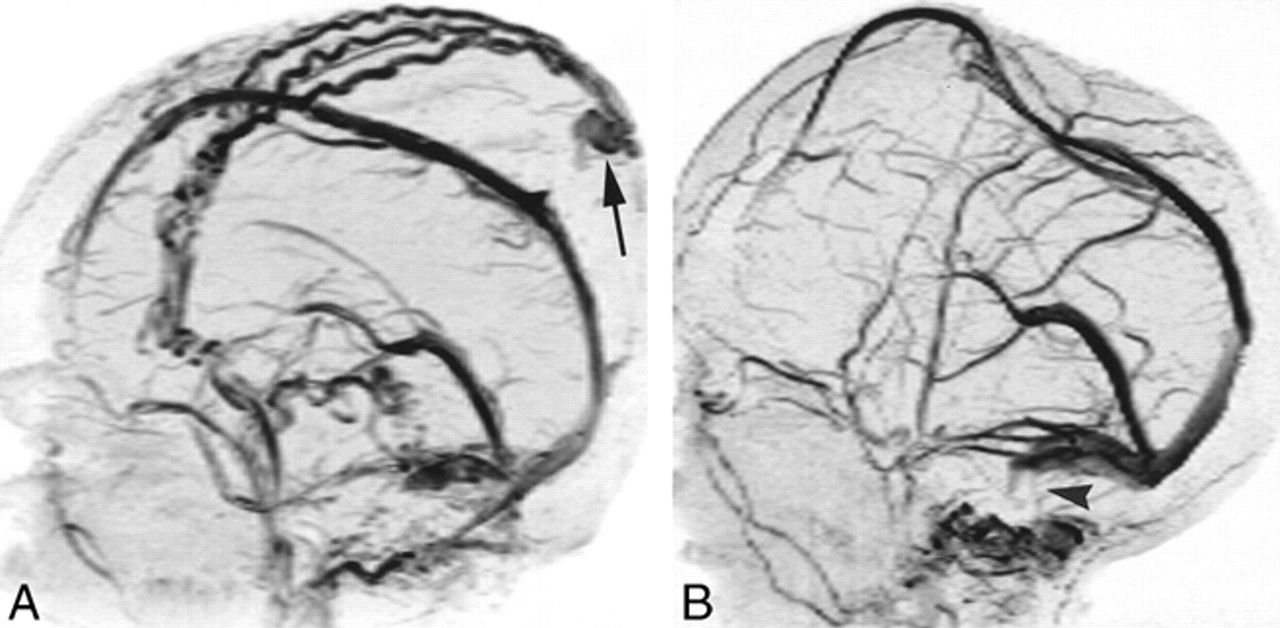
Dural venous sinus stenosis at 2D-TOF MR venography in two patients with AROP.
A, In one patient, lateral projection shows occlusion or high-grade stenosis of both sigmoid sinuses/jugular bulbs. Note prominent scalp collateral venous flow and sinus pericranii (arrow).
B, In another patient (same child as in fig 4A), lateral projection shows bilateral sigmoid sinus stenosis or occlusion (arrowhead). Note superior displacement of superior sagittal sinus by protruding brain.

Cephaloceles, nonsurgical.
A, AROP. Sagittal T1-weighted image shows brain protruding upward into the anterior fontanel as a result of limited intracranial volume. The superior sagittal sinus is superiorly displaced (See fig 3B). Note tonsillar ectopia.
B, AROP. Axial T2-weighted FSE image shows bilateral meningoceles at the craniocervical junction (arrows).
C, ADOP I. Sagittal T1-weighted image shows brain protruding downward through the foramen ovale (arrow). Note scalloping of the floor of the anterior cranial fossa.

Acquired cephaloceles, surgical.
A, AROP. Axial T2-weighted FSE image. Bilateral decompressive craniectomies have resulted in a “Mickey Mouse”-like herniation of brain into and through the calvaria.
B, AROP. Axial T2-weighted FSE image. A posterior fossa decompressive craniectomy was complicated by a cephalocele. Cerebellar tissue, CSF, and meninges protrude through the surgical defect.
Features seen most frequently, or exclusively, in AROP included stenoses of the ICA (Fig 6) and the vertebral artery (Fig 7), expansion of the subarachnoid spaces, brain atrophy, and extramedullary hematopoiesis (Fig 8). Despite the high prevalence of arterial stenosis, no cerebral cortical infarcts were observed in patients with AROP.
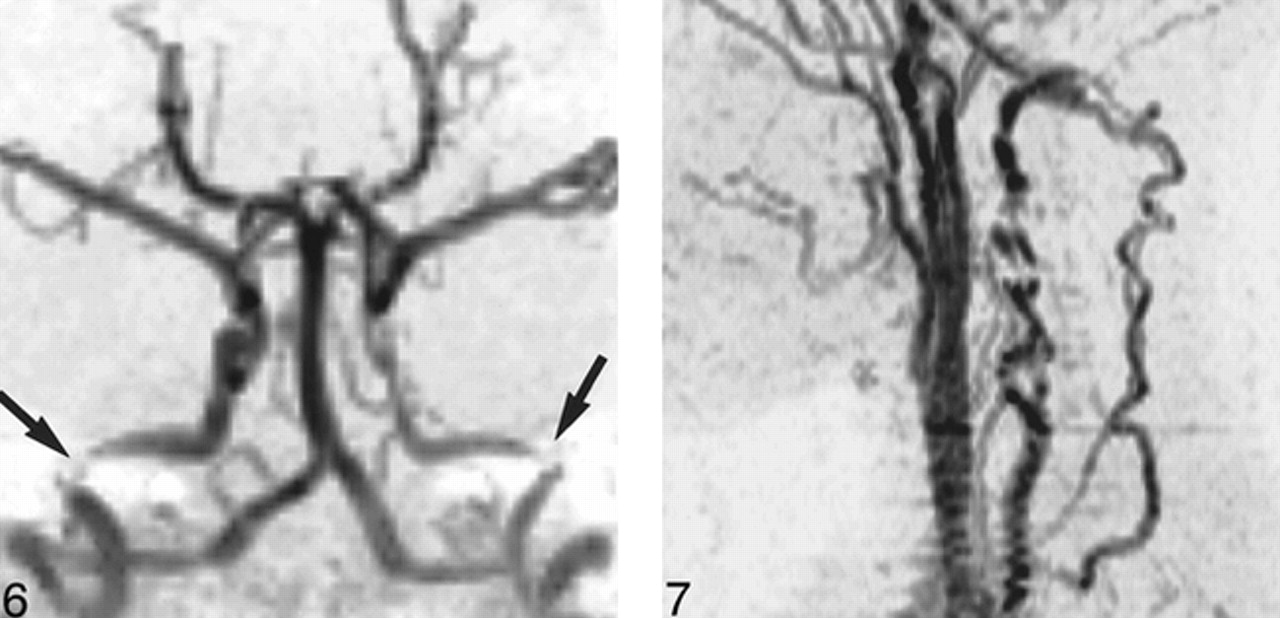
AROP. 3D-TOF MR angiogram, anteroposterior projection, shows bilateral petrous ICA stenosis (arrows), with more diffuse stenosis also evident in the left ICA.
fig 7. AROP. 2D-TOF MR angiogram, left lateral projection, shows segmental vertebral artery stenoses with collateral flow in the ascending cervical arteries, anastomosing with the vertebral arteries above the level of the most cranial transverse foramen.

Extramedullary hematopoiesis, AROP.
A, Axial contrast-enhanced T1-weighted image shows homogeneously enhancing extraaxial tissue along the parietal convexities and falx cerebri.
B, Right lateral projection of 99mTc-sulfur colloid study shows uptake in these deposits of extramedullary hematopoiesis.
Discussion
In this study, the greatest number of observed abnormalities occurred in patients with AROP. This reflects the larger number of these cases in our series, but also the greater severity of osseous abnormalities in this form of osteopetrosis. AROP may be considered the worst-case scenario for osteopetrosis. Both calvaria and skull base are severely involved. The prominent calvarial thickening with limitation of intracranial volume in AROP and ADOP I may account for certain findings common to both types (tonsillar herniation, optic nerve sheath dilatation, and cephaloceles). It is unclear why some features potentially attributable to increased intracranial pressure and limited intracranial volume (eg, empty sella, inner table remodeling) are seen in ADOP I and not AROP.
Bone thickening is associated with stenoses of the optic canal, ICA, and vertebral artery in AROP but not in ADOP I or II. In ADOP I, skull base involvement is less pronounced than calvarial involvement. In ADOP II, the skull base is more severely involved. However, in both types, the disease may have its clinical onset after the arterial and neural foramina have reached their adult size; hence, narrowing of the foramina and canals may not occur. Stenosis of the petrous carotid canals has been found on CT scans in patients with AROP, but no correlation between severity of carotid canal stenosis and age was observed in these patients (4). It is surprising that the patients with AROP experienced no ischemic complications from their severe arterial and venous occlusive disease. The physical inactivity of these children (a result of their anemia, large, heavy heads, and propensity for fractures) may prevent drastic challenges to their cardiac output and brain perfusion. Follow-up of the patients with AROP in this study should further establish the long-term consequences of vascular stenoses. Ventriculomegaly and subarachnoid space enlargement without brain atrophy seen in some patients with AROP and ADOP I may be the result of venous hypertension and compromised CSF resorption, or a tight foramen magnum caused by the combined effects of tonsillar herniation and bone thickening (see Fig 1).
AROP and ADOP II shared a high rate of optic nerve atrophy and stenosis, whereas AROP and ADOP I shared a high rate of optic nerve sheath dilatation. These observations probably reflect the different patterns of skull involvement (calvarial versus skull base) in the two adult forms of the disease. Optic nerve sheath dilatation is probably a manifestation of elevated intracranial pressure rather than of optic canal stenosis. Optic atrophy and optic canal stenosis were not seen in most patients with ADOP I, in which calvarial involvement predominates over skull base involvement. Conversely, in AROP and ADOP II, it is probably optic canal stenosis associated with thickening of the skull base that contributes to optic nerve atrophy, either through optic nerve or ophthalmic artery compression. Middle ear fluid may be a manifestation of eustachian tube stenosis in AROP and IOP, but its frequency in these groups may also reflect the younger age of these patients relative to those with ADOP.
We noted brain herniations (cephaloceles, Fig 5) in all the patients with AROP who had undergone craniotomy. Craniotomies in these patients, most often performed in an attempt to decompress the intracranial vault or optic nerves, are difficult because of hardened vascular bone, and the risk/benefit in the individual candidate should be contemplated carefully.
The large difference in age between patients with AROP and ADOP, and the more complete bony replacement of axial and appendicular marrow spaces seen in AROP compared with ADOP, may explain why extramedullary hematopoiesis is seen in the former but not the latter.
Conclusion
Two forms of osteopetrosis, AROP and ADOP I, share a high rate of occurrence of some MR imaging manifestations, particularly those that relate to calvarial thickening and the resultant limitation of intracranial volume. However, some features are unique to each of the various types of osteopetrosis. The differences in patterns of cranial/intracranial disease probably reflect the differing patterns of involvement of the skull in the variants of osteopetrosis. Since our institution is a major referral center for this disease, the catalog of disease manifestations we have described may be biased by more severe examples. Nonetheless, we hope that our experience sheds light on expected manifestations of the more common forms of this disease.
Footnotes
1 Supported by GCRC Grant RR M01-0170.
↵2 Address reprint requests to Joel K. Curé, MD, Department of Radiology, Medical University of South Carolina, 171 Ashley Ave, Charleston, SC 29425.
- Received September 13, 1999.
- Accepted after revision January 12, 2000.
- Copyright © American Society of Neuroradiology
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Haemodynamic stroke in a rare adult presentation of osteopetrosis
- Magnetic resonance findings in a Cavalier King Charles spaniel with osteopetrosis, Chiari-like malformation and syringomyelia
- Dural calcification and calvarial hyperostosis: a rare cause of obstructive hydrocephalus in 'malignant' osteopetrosis
- Optic Canal Narrowing in Hunter Syndrome (Mucopolysaccharidosis II)
- Spontaneous Optic Nerve Compression in the Osteopetrotic (op/op) Mouse: A Novel Model of Myelination Failure
- The angel in the marble.