
Abstract
BACKGROUND AND PURPOSE: Malformations of cerebral cortical development are common anomalies of the brain, typically causing developmental delay or seizures that are classically thought to begin in childhood. We present clinical and MR imaging data of 16 patients with cortical malformations in whom evidence of neurologic dysfunction was first noted in adulthood, and attempt to determine whether these malformations had any differentiating features from those presenting in childhood.
METHODS: Imaging studies and clinical records of 16 patients with adult-onset neurologic dysfunction were reviewed retrospectively. The patients ranged in age from 17 to 64 years (mean age, 35 years) at the time of imaging. Imaging findings were correlated with seizure history.
RESULTS: Fourteen patients had subependymal heterotopia (seven women, seven men), and two patients had closed-lip schizencephalies. Eleven patients had epilepsy, with age of onset ranging from 14 to 45 years (mean age, 22 years); four of them were successfully controlled by medication. The remaining five patients had no seizure disorder. All patients, except one, had normal intelligence. The bilaterality or multiplicity of location of heterotopias was not associated with the presence or absence of seizures, seizure frequency, or electroencephalographic results.
CONCLUSION: Subependymal heterotopia and small closed-lip schizencephaly may have minor clinical manifestations that are not evident until adulthood, or may, occasionally, never cause neurologic signs or symptoms whatsoever.
Malformations of cerebral cortical development are a fairly common developmental anomaly of the brain. Patients with these malformations typically manifest developmental delay, epilepsy, or focal neurologic deficits that present in the first decade of life (1–8). The experience in our practice has suggested that some patients with malformations of cortical development may not develop any neurologic manifestations until the second decade of life or later. Therefore, we undertook a retrospective study of patients in whom clinical manifestations were first noted in adulthood to determine whether these malformations had any specific differentiating features.
Methods
A retrospective review of imaging studies of patients with malformations of cortical development evaluated at our center over the past 10 years yielded 16 patients (nine women and seven men) in whom neither seizures nor other neurologic dysfunction was detected before the age of 14 years. At the time of imaging, the patients ranged in age from 17 to 64 years, with an average age of 35 years. Cortical malformations were discovered during imaging. Patients were referred to our institution because of seizures (three, new-onset; eight, preexisting), transient aphasia (one), aberrant behavior (one), headaches (one), screening for metastasis from lung cancer (one), or because of a patient's desire to know whether her children's neurologic difficulties were genetically determined (one). Clinical histories were reviewed with specific attention to the following: presence or absence of seizures; type of seizures; age at seizure onset; cognitive or motor deficits; antenatal and birth history; developmental and family history; and electroencephalographic (EEG) results. Antenatal and developmental histories were obtained from patients, and, therefore, were incomplete or imprecise. Extracranial EEG examinations using the International 10–20 system were performed in 12 patients, including one studied with subdural electrodes and one with intraoperative EEG.
Diagnosis of cortical malformations was made on the basis of neuroimaging. Images were reviewed by the authors, and evaluated for the type of cortical malformation and the presence of associated brain abnormalities. In cases of subependymal heterotopia (SEH), the location and number of heterotopia, the size of the largest heterotopic nodule, the presence or absence of the adjacent ventricle enlargement, and thinning or hypogenesis of the corpus callosum were assessed. The heterotopia (singular form, heterotopion) were recorded as “multiple” if they numbered more than five, and “few” if there were fewer than five. Heterotopia were divided further as diffuse, contiguous, or focal. Diffuse heterotopia were defined as bilateral and involving most of the ependymal surface of the lateral ventricles with contiguous nodules. We arbitrarily classified contiguous heterotopia as contiguous nodules that did not involve the entire wall of the lateral ventricles. Focal heterotopia were defined as single or few isolated nodules without contiguity. MR images of patients with schizencephaly were evaluated with special attention to the location, size, and characteristics of clefts. Clefts were classified as closed-lip schizencephaly if the gray matter—lined walls of the cleft were in apposition at all points in more than one plane.
MR studies were performed on a variety of scanners with a number of different techniques, and an equal number of patients were referred to our institution from outlying hospitals as those patients who came directly to our institution. Axial T1- and T2-weighted images and sagittal T1-weighted images were obtained in all patients. Coronal-spoiled gradient-echo images were obtained in three patients. Section thickness varied according to the imaging protocols previously used at each institution, ranging from 1.5 to 7 mm. The number of phase-encoding iterations varied from 128 to 256. The presence or absence and type and severity of seizures, seizure-onset age, and EEG results were correlated with the bilaterality or multiplicity and location and size of heterotopia as well as associated brain abnormalities. Also, the location of any enlarged portion of lateral ventricles and its relation to the site of heterotopia was recorded.
Results
The 16 patients with cortical malformations in whom evidence of focal neurologic dysfunction was absent or first noted in adulthood included 14 with SEH and two with schizencephaly. All results are compiled in Table 1–2, and imaging findings of five patients are shown in Figures 1–5⇓⇓⇓⇓.
Clinical findings in 14 patients with SEH (patients 1–12) or schizencephaly (patients 13–14)
Imaging findings in 12 patients with SEH

Patient 3. Diffuse subependymal heterotopia.
A, Sagittal spin-echo (550/15 [TR/TE]) image shows multiple ovoid subependymal nodules isointense to cortical gray matter, protruding into the lateral ventricle.
B, Axial spin-echo (2500/80) image shows that diffuse heterotopia, lining the lateral walls of the lateral ventricles, remain isointense to gray matter.
C, Midline sagittal spin-echo (550/15) image shows the diffusely thin body of the corpus callosum and a large posterior fossa cyst.

Patient 7. Heterotopia involving the frontal horn. Axial spin-echo (2500/80) image shows subependymal heterotopia associated with the dilated left frontal horn.fig 3. Patient 1. Contiguous subependymal heterotopia.
A, Axial spin-echo (2500/80) image shows multiple contiguous nodules lining the occipital homs and trigones.
B, Sagittal fast spin-echo (3500/102) image reveals the thin splenium.

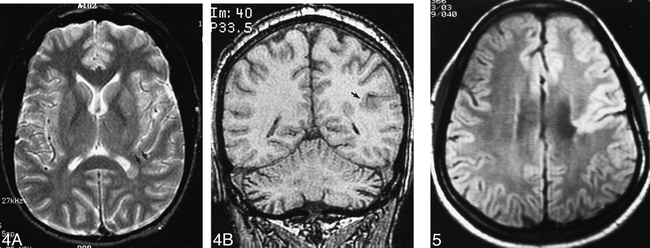
Single heterotopion. Axial spin-echo (2500/80) (A) and coronal 3D FT gradient-echo (35/7 [𝛉 = 35°]) (B) images show that the single heterotopion is associated with deep infolding of the adjacent cortex (arrows).fig 5. Patient 15. Closed-lip schizencephaly. Axial spin-echo (2500/30) image shows small closed-lip schizencephaly involving the left central fissure and decreased white matter volume of the ipsilateral hemisphere
Clinical manifestations
Average age at the time of imaging of 14 patients (seven men; seven women) with SEH was 35 years (range 17–50). Two women with schizencephaly were 22 and 64 years old. Seizure disorders were present in 11 (78%) patients including 10 (83%) with SEH and one (50%) with schizencephaly. Age at seizure onset ranged from 14 to 45 years (mean age = 21.6 years). Three patients had no seizure history. Patient 4, aged 50 years, requested imaging in an attempt to determine whether the disorders in her children were determined genetically: her son had died in childhood with a diagnosis of Sturge-Weber syndrome, and her other two surviving young-adult children have Sjögren's syndrome. Patient 6, aged 41 years, underwent imaging because of recent episodes of transient aphasia, which were suspected to be manifestations of transient ischemia. Patient 13, aged 33 years, was imaged because of a change in the character of her migraine headaches. Patient 14, aged 46 years, was imaged because of episodes of bizarre behavior, suspected to be manifestations of a midlife crisis. Patient 16, after diagnosis of lung cancer at age 64, underwent imaging to rule out brain metastasis; later questioning revealed that he had suffered from a mild hemiparesis since childhood, attributed to infection by poliomyelitis. Seven of the eleven patients with seizures had their initial seizure during or after their third decade of life.
Complex partial seizures were the most common type, affecting six (55%) of the 11 epileptic patients; five eventually developed generalized tonic clonic seizure (GTCS). Focal motor seizures were diagnosed in two patients, including one with schizencephaly who had GTCS also. Three patients manifested GTCS only. The seizures in seven patients were refractory to treatment by two or more antiepileptic drugs. Four (36%) patients with medically controlled epilepsy had only one type of seizure (three, GTCS; one, focal motor seizures). All with partial complex seizures were difficult to control medically.
EEG results were available from 11 patients (10 with SEH, one with schizencephaly). Six had normal EEGs by report, including four with seizure disorders. In the five patients with abnormal EEGs, the most consistent abnormalities were spike and wave discharges. Two patients had abnormal slow waves bilaterally also. In three patients (two of whom had bilateral SEH), EEG abnormalities were bilateral and not localized. Two patients showed unilateral EEG abnormalities in the hemisphere containing heterotopia; one (patient 10) had sharp waves diffusely in the right hemisphere, whereas the other had localizing spikes and waves in the left temporal lobe (patient 11).
All except one patient (patient 8) in this study had normal development, normal childhood behavior, normal intelligence, and normal, successful lives. Our subjects included college graduates, a high school teacher, an attorney, a financial planner, and a computer programmer. Patient 8 had a family history of Hurler's disease, and had low intelligence, as did his two sisters. He showed slow development, and was not able to walk. Imaging revealed he had small open-lip schizencephaly in the left frontal lobe and left diffuse SEH. Both patients with schizencephaly had mild to moderate hemiparesis on the side contralateral to schizencephaly. The other 13 patients had normal motor function.
Family histories were available in 15 patients (Table 1). No patient in our group had first-degree relatives with a seizure disorder. Patient 5 had a maternal great-aunt with a seizure disorder. Patient 12 had a son with a static motor deficit. As mentioned previously, patient 5 had a son who had died with a diagnosis of Sturge-Weber syndrome and two daughters with Sjögren's syndrome.
Imaging findings
Neuroimaging revealed 14 cases of SEH and two of schizencephaly. In cases of SEH, MR imaging revealed isointense heterotopic tissue to gray matter on all pulse sequences in the subependymal area of the lateral ventricles (Fig 1). The third and fourth ventricles were not involved. The SEH were bilateral in six (four women) and unilateral in eight (three women) patients. Four (two women) with bilateral heterotopia had contiguous multiple heterotopic nodules; they were distributed diffusely along the walls of both lateral ventricles in two, and located along the posterior aspects of the lateral ventricles in the other two. Of the eight patients with unilateral SEH, five (62%) had nodules on the left side; three had multiple contiguous nodules, three had two or three nodules, and the remaining two had a single nodule. The posterior aspects of the lateral ventricles (occipital horns and trigones) were involved in 10 patients (71%), whereas the frontal horns were involved in three (21%). The thickness of nodules varied from 3 to 10 mm; size did not correlate with multiplicity or bilaterality. Neither the location, multiplicity, nor bilaterality of the heterotopia correlated with the presence or absence, severity, or type of seizures or the type of EEG abnormalities.
One or both lateral ventricles were dilated focally or diffusely in nine patients (64%); the enlarged portion of the lateral ventricle was concordant with the sites of heterotopic nodules in eight patients (88%), including one who had diffuse symmetric bilateral heterotopias and diffuse dilation of both lateral ventricles. One patient had multiple heterotopias on the left side, but the right lateral ventricle was enlarged. Three patients without ventricle enlargement had one to three SEH.
Thinning or hypogenesis of the corpus callosum was detected in four (29%) patients, including one with hypogenesis of the posterior body and splenium, two with a thin splenium, and one with a diffusely thin body. All had multiple contiguous nodules (three, bilateral; 1, unilateral), close to the abnormal portion of corpus callosum. Other associated anomalies included deep infolding of the cortex (two patients), a large posterior fossa cyst with vermian hypoplasia (one patient), and a large cavum septum pellucidum (one patient). Patient 8 had subcortical heterotopia and small schizencephalies in the ipsilateral temporal and frontal lobes. Patient 11 had a small subcortical heterotopia in the contralateral corona radiata. Patient 2 had cerebellar atrophy, which was thought to be secondary to phenytoin therapy.
Patient 15 and 16 had closed-lip schizencephalies in the left and right central fissures, respectively, and the white matter volume within the ipsilateral hemispheres was decreased. Neither patient had associated intracranial anomalies, although Patient 16, aged 64, had focal ischemic changes, presumably attributable to microangiopathy.
In all but patient 8, who had subcortical and subependymal heterotopia in the temporal lobe, the gross appearance of hippocampi looked normal; this included the hippocampal appearance of three patients imaged with contiguous 1.5-mm sections.
Discussion
Malformations of the human neocortex have been recognized for more than 100 years (9), and were considered to be extremely rare disorders that were seen exclusively in individuals with severe intellectual and neurologic disabilities. Recent advances in neuroimaging have revealed that cortical malformations are much more common than were expected previously, and not uncommonly are found in children with developmental delay, cognitive deficit, and epilepsy (5–7, 10–14). Our series supports previous investigations suggesting that some patients with less severe cortical malformations, such as gray matter heterotopia (6, 14–16) or schizencephaly (7), have normal intelligence and development; seizures may not develop until the second decade of life. Moreover, some patients have no neurologic signs or symptoms whatsoever.
Gray matter heterotopia are aggregates of abnormally located neurons that result from an arrest of radial migration. Heterotopia have been associated with a wide variety of genetic, vascular, and environmental causes (17, 18). The exact pathophysiologic process of the migration arrest has not been established. Several explanations have been proposed, such as a damage to the radial glial fibers (1), a deficiency of the specific surface molecules necessary for the migration of neuroblasts (17), or a premature transformation of the radial glial cells into astrocytes (18).
Gray matter heterotopia can be classified as subependymal, focal subcortical, and band heterotopia. Although these classifications are anatomic, morphologic, and, to a certain extent, artificial divisions, each of them recently has been considered as distinct clinicoradiologic entities (2, 6, 7). SEH are thought to be the most common form of gray matter heterotopia, and have the fewest clinical manifestations despite their macroscopic abnormalities revealed by neuroimaging. Our findings support the concept that SEH are the most benign form of heterotopia. Not only do patients with SEH have normal intelligence and developmental milestones, they may not develop epilepsy until their fourth decade of life, if at all, as with our patient 4.
Our review of the literature indicates that SEH are more likely to be bilateral than unilateral. Also, it has been stressed SEH occur more often in women, and have a frequent familial component, including a family history for epilepsy (6, 15, 16, 19, 20). These features suggest a different etiology for bilateral and unilateral SEH (21). Genetic factors seem to play a role in the development of familial SEH; a recent linkage study showed the familial type to be closely connected to a dominant gene located in the Xq28 region (22). These familial heterotopia appear to be bilateral, diffuse, and contiguous. In contrast, unilateral cases do not show a strong female or a familial prevalence (21).
In our series, unilateral heterotopia were more common than bilateral, and there was no significant predominance in women for either unilateral or bilateral cases. Nonetheless, both patients with bilateral diffuse heterotopia, involving the entire lateral ventricles, were women. Only one patient had a positive family history for seizures, although it was not confirmed whether patients had relatives with SEH. Patient 5, who had only a few nodules in the occipital horns of the lateral ventricles, had a maternal great aunt with epilepsy. It is interesting that patient 4, a high school teacher with bilateral diffuse SEH, had a son who died at age 15 months with a clinical diagnosis of Sturge-Weber syndrome (no neuroradiologic or pathologic studies were performed) and two daughters with Sjögren's syndrome. It is interesting to speculate that the son may have had the lethal form of X-linked SEH that is seen in affected men (16, 23). Affected men have early-onset refractory epilepsy, developmental delay, and neurologic signs and symptoms. There is no established theory regarding the possible relationship between maternal heterotopia and filial Sjögren's syndrome.
Epilepsy was the only clinical manifestation in our patients with SEH. Of the four patients without seizures, one had no clinical symptoms, one was evaluated for migraine headaches, and the other two had episodes that were judged not to be epileptic in nature. Both had normal EEGs, and the one with episodes of transient aphasia presently is being monitored in order to determine if the aphasia has a vascular etiology (19). In those patients with epilepsy, the seizure pattern is variable. Most affected individuals have medically refractory partial epilepsy as well as generalized epilepsy. Eighty percent of our patients with heterotopia and epilepsy had GTCS, including five with both complex partial and generalized epilepsy. The three patients with GTCS were controlled completely by medical therapy, whereas the complex partial seizures in the other five patients were refractory to antiepileptic drugs. This observation is consistent with the findings of other authors (21).
There is no established theory about cause of the seizures with heterotopia. Some electrophysiologic studies have demonstrated intrinsic epileptic discharges from band heterotopia (23) and subcortical heterotopia (24). In a recent report, depth electrodes did not record epileptic activity from a subependymal nodule, but subtotal removal of the heterotopic nodule and adjacent (histologically normal) hippocampus, rendered the patient almost seizure-free (25). In another report, depth electrodes demonstrated independent epileptic discharges from both the hippocampus and a subependymal heterotopion in a patient with partial epilepsy (16). These reports suggest that SEH contribute to, but are not solely responsible for, the seizures in affected patients. Moreover, it is estimated that about 15% of patients with hippocampal sclerosis have associated malformations of cortical development (26, 27), the most common of which is SEH (26). Intraoperative EEG in one patient showed epileptic discharges from the temporal lobe, and the epileptiform activity ceased after temporal lobectomy. Nonetheless, most authors report poor outcome after temporal lobe resection for SEH and electroclinical features suggestive of temporal lobe epilepsy, even when the patients have histologically proved hippocampal sclerosis (25). Poor outcome also is reported after SEH resection, even in the presence of the relatively well-delineated epileptogenic area (16). These features raise the question of whether subependymal nodules are the entirety of the dysgenesis or merely the visible part of a more widespread developmental abnormality that is mostly beyond the limit of MR resolution.
SEH can be associated with other abnormalities including hydrocephalus, agenesis of the corpus callosum, and polymicrogyria; the presence of associated anomalies seems to portend an unfavorable clinical course in most series. In our series, four of seven patients with diffuse or contiguous multiple heterotopia had hypogenesis of the corpus callosum. Two patients had deep cortical infolding adjacent to heterotopia; this association has been reported previously (6). One patient in our series with bilateral diffuse heterotopia (patient 3) had a large posterior fossa CSF collection. This association also has been documented. In one series, five of seven patients with bilateral SEH had mega cisternae magna (21). One interesting finding in our series is localized dilatation of the lateral ventricles, generally congruent with heterotopia sites. This focal dilatation is, perhaps, a manifestation of local white matter hypogenesis.
The finding of schizencephaly in two patients imaged relatively late in life is somewhat surprising, as schizencephaly is generally considered a severe malformation that is associated with significant motor and intellectual disabilities. Recent studies, however, have reported that severity of motor and mental dysfunction is closely related to the size and location of the clefts, indicating a wide spectrum of clinical presentation (7, 28). Most patients have some degree of motor impairment because the perirolandic regions are involved commonly; such was the case in both of our patients. Our patient 16, aged 64 years, had only mild longstanding hemiparesis as the sole clinical manifestation. Her paresis was attributed initially to poliomyelitis, which had been diagnosed during her infancy. The finding of schizencephaly in the hemisphere contralateral to the hemiparesis, however, challenged this initial diagnosis. Although we acknowledge that most patients with schizencephaly have seizure disorders that are well controlled by medication (7, 28), we speculate that undiagnosed schizencephalies with little or no neurologic impairment are more common than generally accepted.
Conclusion
We conclude that malformations of cortical development may have mild manifestations, which may not result in any neurologic abnormalities until adulthood, and, in some cases, may not cause any neurologic manifestations whatsoever. Most of these patients have SEH. The number and location of these heterotopia did not correlate with the presence or absence of epilepsy. Surprisingly, patients with closed-lip schizencephalies may not be diagnosed until adulthood; these individuals have normal intelligence and mild contralateral hemiparesis. Epilepsy may or may not be present. It appears that malformations of cortical development should be considered within the differential diagnosis of patients presenting at any age.
Footnotes
↵1 Address reprint requests to A. James Barkovich, MD, Neuroradiology, Rm L-371, UCSF, 505 Parnassus Ave., San Francisco, CA 94143-0628.
References
- Received August 27, 1998.
- Copyright © American Society of Neuroradiology
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.