
Abstract
Summary: A 4-month-old boy with polydactyly and bifid epiglottis was found to have a large sellar and suprasellar mass. When the diagnosis of Pallister-Hall syndrome was made, conservative management was elected. When the patient was 2 years old, the tumor had grown proportionally with the patient, and he was developing appropriately. Although rare, this entity is important to recognize not only for clinical diagnosis but also for appropriate management and genetic counseling.
In 1980, Hall and colleagues first described a syndrome characterized by congenital hypothalamic “hamartoblastoma,” hypopituitarism, imperforate anus, postaxial polydactyly, and various visceral anomalies (1). Since their report, cases of more mildly affected individuals with Pallister-Hall syndrome have been reported, including cases of asymptomatic individuals (2, 3). Familial cases with an autosomal dominant inheritance pattern have been described also in the genetics literature (2, 4). We report the imaging findings in a case of Pallister-Hall syndrome that has been followed successfully and conservatively for 2 years.
Case Report
A 4-month-old African American male patient with sickle cell trait, who was being evaluated for poor weight gain and frequent regurgitation, was found to have a bifid epiglottis (Fig 1) and tracheomalacia.

Single image from laryngoscopy demonstrates large midline cleft (black arrow) through the epiglottis (white arrows).
fig 2. Anteroposterior conventional radiograph of left hand shows polydactyly with hypoplastic accessory metacarpal, proximal, and mid phalange between fourth and fifth digits.
The patient was the product of an uncomplicated pregnancy with normal spontaneous vaginal delivery at 37 weeks. There was no known maternal exposure to drugs, alcohol, or other teratogens. Apgar scores were 7 and 8 at 1 and 5 minutes, respectively. At birth, his weight was 2865 g (50%), length was 48.5 cm (60%), and head circumference was 31.5 cm (<5%).
Physical examination at birth revealed a flat, broad nasal root and upturned nose. Facial exam was otherwise unremarkable. There was insertional hexadactyly of the left hand (Fig 2) and post-axial skin tags on the ulnar aspect of the right fifth and left sixth digits. The distal phalanges of the hands were short with hypoplastic fingernails.
At the time of presentation, the patient was at approximately the third percentile for height and weight. Because of concern for possible midline abnormalities, an MR image of the brain was obtained that revealed a large sellar and suprasellar mass (Figs 3–4). Renal and cardiac echograms were normal. Subsequent endocrinologic workup at 2 years old revealed normal serum T4, thyrotropin, cortisol, and corticotropin levels. Nonetheless, growth hormone, IG1, and insulin-like growth factor binding protein levels were low, consistent with partial growth hormone deficiency. In addition, follicle-stimulating hormone and luteinizing hormone levels were low. Chromosomal analysis revealed 46 X, Y karyotype without evidence of cytogenetic abnormalities. Family history was noncontributory and brain MR images of the patient's parents were unremarkable.

Sagittal T1-weighted (580/14/2 [TR/TE/excitations]) MR image demonstrates large sellar and suprasellar mass extending over and through dorsum sella and into pontine cistern. The infundibulum is anteriorly displaced and pituitary (arrow) is compressed. Posterior displacement of pons and midbrain with superior displacement of third ventricle is visible.
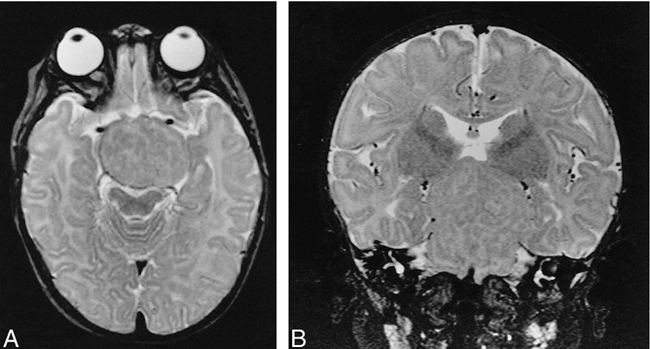
A and B, Axial (A) and coronal (B) T2-weighted (2500/90/1) MR images show heterogeneous appearance to tumor with mixed signal intensities equivalent to white and gray matter of remaining normal brain.
Neurosurgical resection of the mass was scheduled but cancelled after the diagnosis of Pallister-Hall syndrome was established. The patient demonstrated mildly delayed mental development at 13 months old when the Bayley Scales of Infant Development were applied. Motor development was delayed mildly to moderately with the patient walking at 18 months old. Follow-up MR imaging of the brain at 2 years old showed that the hypothalamic hamartoma grew proportionately with the patient. The patient had no visual or auditory symptoms. At 26 months old, the patient was 5% in height, 50% in weight, and 40% in head circumference; however, with the aid of occupational and physical therapy, the patient appeared to be developing more appropriately. He was noted to have a microphallus with normally descended testes.
Discussion
The primary feature of Pallister-Hall syndrome is the hypothalamic hamartoma. Other major manifestations of the syndrome include polydactyly, dysplastic nails, bifid epiglottis, imperforate anus, renal anomalies, pituitary dysplasia, and hypopituitarism (5).
In familial cases, Pallister-Hall syndrome has been noted to be inherited in an autosomal dominant pattern with variable expressivity. It has been linked with a mutation to a zinc finger transcription factor gene, GLI3, which resides on chromosome 7p13 in some inherited probands (6, 7). Mutations to the GLI3 gene have been associated also with Greig cephalopolysyndactyly syndrome.
Histologic examination of the hypothalamic lesions in patients who died as neonates demonstrated primitive germinal cells, which indicated a neoplastic potential (1, 8). For this reason, the term hypothalamic hamartoblastoma was assigned initially to these tumors. Subsequent pathologic examination of tumors from older patients with Pallister-Hall syndrome, however, has revealed disordered arrangements of large mature-appearing neurons admixed with astrocytes and minimal white matter (9, 10). The white matter has shown varying degrees of myelination. Because these tumors have a more mature histologic appearance and benign clinical course, they are thought now to be hypothalamic hamartomas. The previous description of undifferentiated germinal cells within these tumors is felt now to represent the hamartomatous equivalent of the neonatal germinal matrix (9).
Disruption of pituitary development from the hypothalamic hamartoma can result in endocrine abnormalities, which were a prominent feature in the initial description of Pallister-Hall syndrome. The main cause of mortality in the first-described cases was acute adrenal insufficiency associated with panhypopituitarism (1). A wide spectrum of pituitary abnormalities has been reported subsequently ranging from asymptomatic individuals to panhypopituitarism (11). Hypothyroidism, microphallus, and cryptorchidism have been reported also.
Craniofacial abnormalities are felt to be secondary to disruption of midline development by the hypothalamic hamartoma. These include a short nose with flat nasal bridge, low-set and posteriorly angulated ears, cleft palate, cleft uvula, buccal frenula, bifid epiglottis, and cleft larynx.
Limb abnormalities include polydactyly, short limbs, syndactyly, and nail dysplasia involving the toes and fingers. Other genitourinary abnormalities described in addition to microphallus and cryptorchidism are renal hypoplasia or agenesis and renal ectopia. Reported congenital heart defects include patent ductus arteriosus, ventricular septal defect, endocardial cushion defect, mitral and aortic valve defects, and proximal aortic coarctation. Pulmonary segmentation anomalies are associated also with Pallister-Hall syndrome (11).
Other conditions with overlapping features include Ellis-van Crevald syndrome (congenital heart defects, polydactyly, multiple frenula, and natal teeth), Smith-Lemli-Opitz syndrome (polydactyly and various CNS anomalies), oral-facial-digital syndrome type VI (autosomal recessive, polydactyly, tongue hamartomas, and cerebellar vermis hypoplasia), Kaufman-McKusick syndrome (autosomal recessive, hydrometrocolpos, polydactyly, and congenital heart defects), and Grieg cephalopolysyndactyly syndrome (autosomal dominant, polydactyly, and craniofacial abnormalities) (3, 11). As previously discussed, the hypothalamic hamartoma of Pallister-Hall syndrome is the key feature in differentiating it from these aforementioned entities.
On MR imaging, the classic hypothalamic (tuber cinereum) hamartoma is noncalcified and nonenhancing, and is homogeneously isointense to gray matter on T1-weighted images, isointense to mildly hyperintense on proton density–weighted images, and often hyperintense on T2-weighted images. These imaging findings are fairly characteristic and are helpful in differentiating the hypothalamic hamartoma from the more common suprasellar lesions such as craniopharyngioma and hypothalamic/opticochiasmatic glioma seen in children. Craniopharyngiomas are usually heterogeneous with cystic and solid enhancing components as well as focal calcifications. The cystic areas have a variable appearance on MR images but often have increased signal on T1-weighted images. Gliomas have variable enhancement, are hypointense to isointense on T1-weighted images, hyperintense on T2-weighted images, and are usually more heterogeneous in appearance than hamartomas. This particular case is interesting both for the unusually large size of the tumor and for the apparent MR findings of prominent white matter tracts coursing throughout the tumor, suggesting a relatively mature histologic course.
The importance of recognizing Pallister-Hall syndrome should be stressed not only for clinical management but also for future genetic counseling. In this case, the proper diagnosis prevented an unnecessary neurosurgical procedure for this patient. Clinical management of these patients should include endocrinologic evaluation of the hypothalamic-pituitary axis, ophthalmologic evaluation with visual-field testing, serial MR imaging for tumor progression, and a dedicated search for associated anomalies (11). It is recommended also that the parents of affected individuals be screened for asymptomatic hypothalamic hamartomas with cranial MR imaging.
Acknowledgments
We gratefully acknowledge the invaluable assistance of Drs. Nancy J. Mendelsohn and Lester Biesecker.
Footnotes
↵1 Address reprint requests to Charles L. Truwit, MD, Department of Neuroradiology, University of Minnesota, Box 292, 420 Delaware St. NE, Minneapolis, MN 55455.
References
- Received December 1, 1998.
- Accepted after revision June 23, 1999.
- Copyright © American Society of Neuroradiology




{kind=link}
{kind=link}
{kind=link}